
Don Wyatt and Dr. Keith A Crandall, Zoology
The ecology, morphology, behavior, and geographical distribution of freshwater crayfish are extremely diverse. The genus Cambarus is the second largest in species richness, containing over 25% of the crayfish diversity in North America. The subgenus jugicambarus contains 21 species. Relationships among the taxa within this group are not well understood. The objectives of this research were to:
1. Determine the relationships among taxa within a group of crayfish, subgenus jugicambarus.
2. Use the phylogenetic relationships among these taxa to make conclusions about the origins of different ecological niches occupied by these taxa.
3. Become familiar with the laboratory and analytical methods required for studies in molecular systematics.
Laboratory Methods and Analyses
DNA was extracted from seven Cambarus species within the subgenus jugicambarus (Table 1), using the method of Crandall et al. (1999). The polymerase chain reaction (PCR) was used to amplify two non-coding mitochondrial gene regions, 16S and 12S rDNA. Amplification and sequencing were done as described in Crandall and Fitzpatrick (1996). The resulting PCR product was verified using 1% agarose gels. PCR products were cleaned using the Gene Clean kit (Bio101). Sequencing reactions were performed using the ABI Big-dye Ready-Reaction kit, following the standard cycle sequencing protocol, but using a quarter the reaction size. Sequences were generated on an ABI 377 automated DNA sequencer and edited using the Sequencher program (Gene Codes Corp. Inc. 1995). Sequences were aligned in ClustalX (Thompson et al. 1997), and adjusted by eye. Table 1. List of species sequenced and their ecological habitats (S = lotic stream, L = lentic ponds, B = burrower, C = cave).
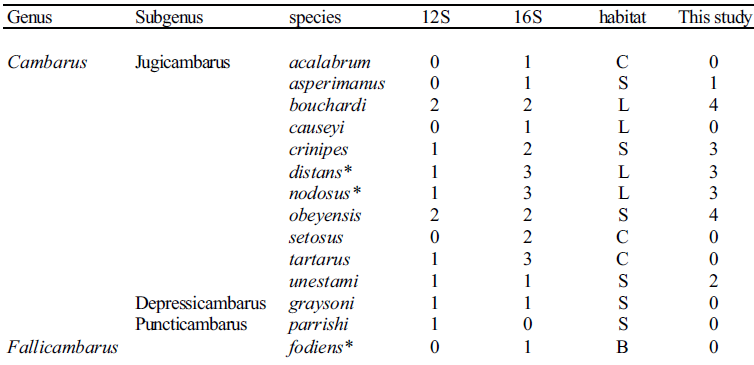
![]()
The computer program ModelTest (Posada and Crandall 1998) was used to determine the most appropriate model of evolution for each gene region. These models were implemented in a likelihood analysis, using PAUP* 4.0 (Swofford 2000). A heuristic search was performed with 100 replicates. Confidence in the nodes was assessed using 500 bootstrap resampling replicates. A partition homogeneity test (Farris et al. 1994) was performed to determine whether data from these two gene regions could be combined, that is whether the phylogenetic signal from these two regions was significantly different or not. Sequences from Barbicambarus cornutus were used as an outgroup, and several other Cambarus sequences were included from the lab.
Results and Discussion
Eight sequences were obtained for 288bp of the 12S region and twelve for 451 bp 16S. An HKY85+G model best explained the12S data (Table 2), while a Tamura-Nei+G model best explained the 16S data.

The partition homogeneity test was not significant (p =1.00), indicating that we could do a phylogenetic analysis of the combined data. For the combined data set, the Tamura-Nei+G model best explained the data. Relationships among sequences for the 16S and combined data sets are shown (Fig. 1a & b). The 12S tree (not shown) had the same topology as the 16S tree, except that there were fewer taxa. There was strong bootstrap support for clades ‘A’ and ‘B’ for the 16S tree. Low bootstrap support for other clades is consistent with the short branch lengths. The short branches are typical of rapid radiation within a species group. There was no support for monophyly of the jugicambarus group or for those species associated with the four different ecological habitats.
This provides evidence for multiple origins of the morphologies associated with these habitats. Field identification of many of these species is difficult, as they are morphologically very similar (within their habitat type). The ID’s of several of the taxa are under question (marked *). Clearly, the results from this study warrant further data collection (some missing taxa) and re-examination of the specimens. Another more variable gene region may also give better resolution among taxa.
References
- Crandall, K.A. and Fitzpatrick, J.F. Jr. (1996) Crayfish Molecular Systematics: Using a Combination of Procedures to Estimate Phylogeny. Systematic Biology 45: 1-26.
- Crandall, K.A., J.W. Fetzner Jr., S.H. Lawler, M. Kinnersley, and C. M. Austin. (1999) Phylogenetic relationships among the Australian and New Zealand genera of freshwater crayfish. Australian Journal of Zoology 47: 199-214.
- Farris, J.S., Källersjö, M., Kluge, A.G. and Bult, C. (1994) Testing significance of incongruence. Cladistics 10: 315-320.
- Posada, D. and K.A. Crandall. (1998) Modeltest: Testing the model of DNA substitution. Bioinformatics 14: 817-818.
- Swofford D. (2000) PAUP* 4.0 Sinauer Associates, Sunderland MA.
- Thompson J.D., Gibson T.J., Plewniak F., Jeanmougin F. and Higgins D.G. (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Research 24: 4876-4882.