
Jesse Rowley and Dr. Gregory Burton, Microbiology
Human Immunodeficiency Virus (HIV), the cause of AIDS, has eluded successful treatment since its discovery in the early 1980s. HIV is a retrovirus that infects and eventually destroys CD4+ T cells. In HIV infected individuals it is believed that during the longest phase of infection (latency) most viral production takes place in and around the germinal centers of secondary lymphoid tissues. Follicular dendritic cells (FDCs) are a unique component of the germinal center, and previous studies done in our laboratory have suggested that FDCs may augment viral replication and/or infection within the germinal center.
Infection is initiated when a virion attaches to and penetrates a T cell. After penetration, viral DNA is produced from viral RNA and becomes integrated into host cell DNA. Integrated HIV (provirus) is transcribed into RNA which will either be translated into viral proteins or packaged into infectious viral particles. Once released from the cell, these particles are capable of infecting surrounding T cells. Also, because HIV is integrated into the cellular genome, as each infected T cell divides, each progeny T cell will contain the provirus. Further, studies have shown that in the presence of FDCs, T cell division is increased. We hypothesized that FDCs potentiate HIV infection by enhancing proliferation of infected T cells, thus increasing the number of provirus containing T cells.
We proposed to test our hypothesis in vitro by using an env deficient virus to cause a single round of infection of a set of CD4+ T cells (Fig. 1). The protein product of the HIV env gene facilitates attachment and penetration of CD4+ T cells. If the env gene is absent in provirus containing T cells, viral particles will be produced that will not be able to infect surrounding T cells. 
The techniques used to produce such a virus are new to our laboratory, and this paper will document the results obtained constructing this virus. Once constructed, infection with an env deficient virus should then allow us to track solely the progeny of infected T cells by eliminating the ability of the virus to infect surrounding T cells. By measuring the number of HIV infected T cells in the presence and absence of FDCs, we should be able to determine if the FDCs affect the proliferation rate of infected T cells.
Plasmids are small circular pieces of DNA that can be introduced into (transfected) and produce proteins in foreign cells. Construction of the env deficient provirus required the isolation and double transfection of the HIV-gpt plasmid which contains an env deficient HIV genome, and the HXB-env plasmid which contains only the env portion of the HIV genome. Combined, these complementary plasmids produce infectious HIV particles containing an env deficient genome. Upon subsequent infection of T cells only the HIV-gpt env deficient genome will be integrated into the host cell DNA, resulting in an env deficient provirus containing T cell.
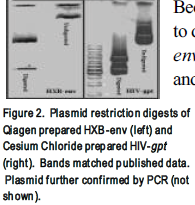
Because there are many different methods for plasmid isolation, we first sought to determine the optimal method of plasmid isolation for the HIV-gpt and HXB-env plasmids. Plasmid containing bacteria were grown overnight, harvested, and the cell membranes lysed, and plasmids were purified by a phenol:chloroform extraction, a cesium chloride density gradient, or a kit available from Qiagen. After isolation, plasmids were quantified by spectrophotometric analysis, digested with restriction enzymes, and/or analyzed by gel electrophoresis or PCR. Figure 2 shows restriction digests of the HXB-env and HIV-gpt plasmids. As determined by gel analysis the, the Qiagen kit produced the purest plasmids (Fig. 2) and was used for all future isolations. The cesium chloride method (Fig. 2) produced plasmids that were almost as pure; however transfection with these plasmids was unsuccessful (data not shown). The phenol:chloroform method produced low purity plasmids (not shown).
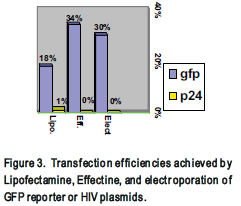
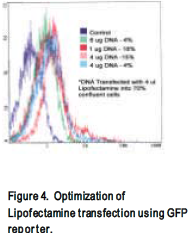
Before attempting transfection with the HIV plasmids, three different transfection methods were optimized. Commercially available Lipofectamine (Gibco) and Effectine (Qiagen) reagents were used to transfect neoplastic 293 cells and electroporation was used to transfect H9 or Jurkat cells. Lipofectamine and Effectine transfections were optimized by varying the amounts of the reagents, DNA, and cell confluency (Fig. 4). Transfection efficiency was determined using a GFP reporter plasmid, which produces a fluorescent protein easily detected by flow cytometer (Fig. 3 and 4).
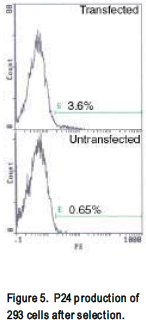

HIV-gpt and HXB-env plasmids were transfected into 293 or H9 cells. Success of transfection was tested by flow cytometer staining for p24 (a product of the HIV-gpt plasmid) and env. Initially, using Lipofectamine, 1% cells stained p24 positive (not shown). After cells were selected for 2 weeks in mycophenolic acid an increase to 3% of p24 producing cells was observed (fig 5). Env production was not detected by flow cytometer. Further, western blot on transfected cell lysate did not indicate any env production had occurred. Neither Env nor p24 production was not detected in electroporated H9s; curiously however, when analyzed by southern blot, a more sensitive assay, the presence of the gag gene (part of the HIV-gpt plasmid) was detected (Fig. 6). Supernatant was harvested from these transfected cells, added to uninfected H9s, and after 3 days the cells were analyzed on flow cytometer and by southern blot. Once again p24 was not detected by flow cytometer, however the presence of gag was detected by southern blot (Fig. 6).
At this point, infectious env-deficient virus, indicated by the presence of gag in infected H9s, has been produced. For our experimental purposes however, we must still produce virus that can be detected by flow cytometry. We believe this may be achieved by increasing transfection efficiencies of the viral plasmids, increasing protein production of transfected cells, applying a more stringent selective agent, or increasing the p24 staining sensitivity. After we have successfully produced an env deficient virus that can be detected by flow cytometer, we will use the virus to infect T cells and measure the effects FDCs have on infected T cell proliferation.