
Steven Woolley and Dr. David McClellan, Zoology
The program TreeSAAP measures the selective influences on several structural and biochemical amino acid properties during phylogenesis (the history of genealogical development) and performs goodness-of-fit and categorical statistical tests.
Calculating the above information by hand is a very laborious task which grows exponentially more difficult as the number of amino acid sequences increase. The basic model for these calculations was developed by Dr. McClellan who wrote the preliminary code for these calculations.1 It was my task to create a robust application to perform the analysis in different scenarios, using a simple interface. The program is called TreeSAAP (Selection on Amino Acid Properties using phylogenetic Trees).
In the past year, I have improved the TreeSAAP program in many areas. Aside from routinely fixing errors that arose, I added several new functions to TreeSAAP. I also changed the graphical user’s interface to make it more intuitive and simple for new users.
One of the first additions to TreeSAAP was the computations for another model developed by Dr. McClellan, called CDM.2 The output from this calculation can be viewed from TreeSAAP or in any text editor.
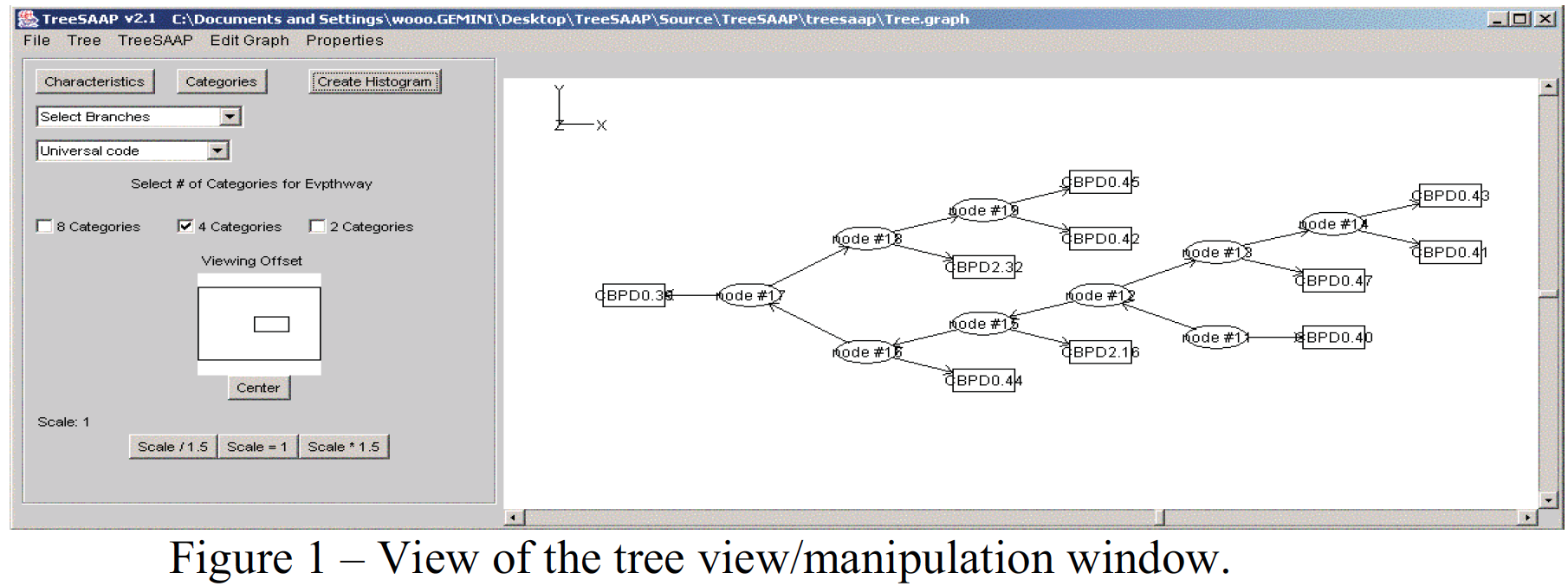
The major addition to the functionality of TreeSAAP was a graphical tree editor/viewer. When analyzing ancestral trees using TreeSAAP, ancestral DNA sequences are calculated using the baseML from the PAML software package.3 In the preliminary version of TreeSAAP these calculated sequences were used only for one calculation and were then discarded. Since calculating these sequences is a very time consuming process, this approach was very inefficient. I needed a method of saving not only these ancestral sequences of DNA but also keeping track of their relationship to the original sequences. The format for the genealogical trees input into TreeSAAP only specifies the positions of the terminal sequences relative to each other but give no information for the internal nodes. In order to store this additional data I choose to use the GML format used to store graphs and networks.4
With the tree structure and data being saved, a new option was available. Firstly, I was able to display and manipulate the tree including all internal branches using the freeware VGJ (Fig. 1).5 With this software the user can manipulate the tree’s structure, and can perform the evaluations on selected parts of the tree. He or she can even create new branches or sequences that can be integrated into the tree. Also through this new development, it is no longer necessary to run the time consuming baseML again, since it stores the data that it generated.
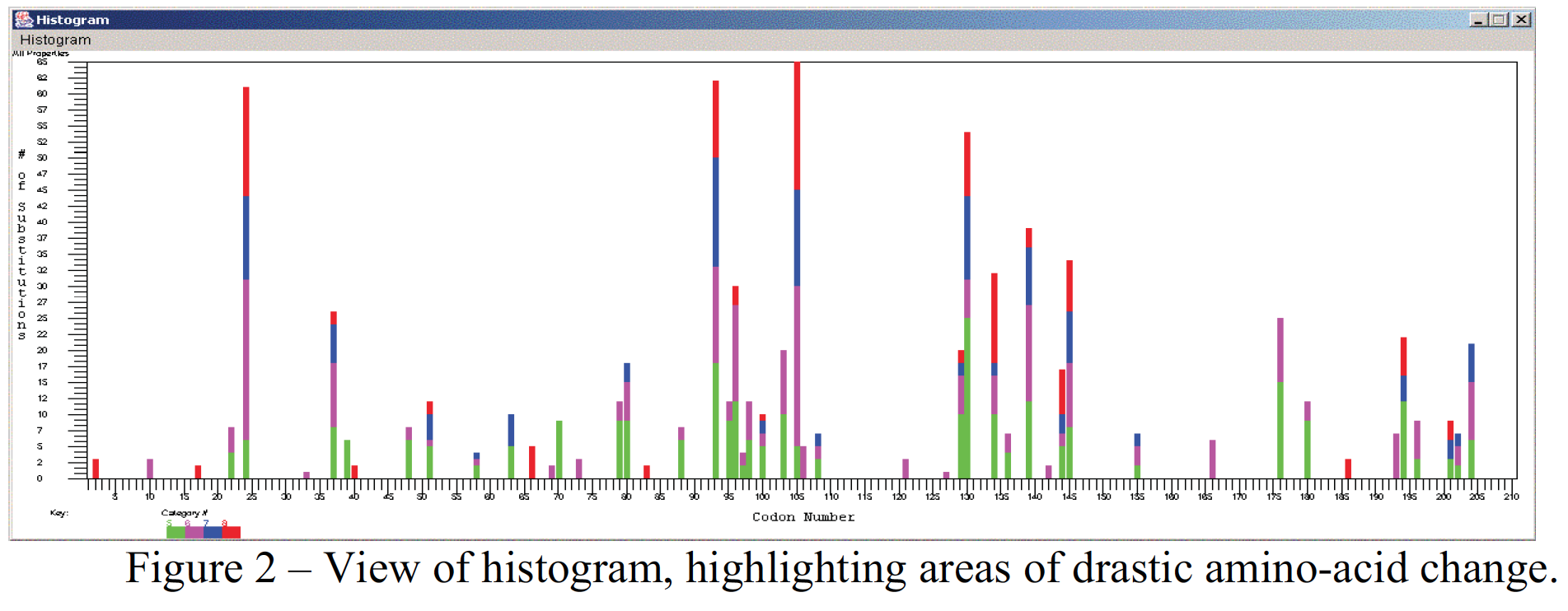
Another goal for the TreeSAAP project was to implement a mechanism that facilitates the interpretation of the data. I choose to add a graphical output to summarize the different amino-acid substitutions, their location, and their relative magnitudes according to the structural and biochemical properties of the amino-acids. The program will now display a summary of this information in a histogram window, when desired (Fig. 2). The data for the histogram can be saved in a comma delimited table format that can be read by any spreadsheet application. This helps the user to pinpoint “hot-spots” in the eventual protein that are changing more than others.
References
- McClellan, D. A. and McCracken,K.G. (2001) Estimating the influence of selection on the variable amino acid sites of the cytochrome b protein functional domains. Mol. Biol. Evol. 18:917-925.
- McClellan, D. A. (2000) The Phylogenetic Utility of the Codon-Degeneracy Model. J. Mol. Evol. 51:185-193
- Yang, Z. (1996) Phylogenetic analysis using parsimony and likelihood methods. J. Mol. Evol. 42:294-307.
- Himsolt, M. GML: A portable Graph File Format http://www.infosun.fmi.uni-passau.de/Graphlet/GML/gml-tr.html
- Drawing Graphs with VGJ http://www.eng.auburn.edu/department/cse/research/graph_drawing/graph_drawing.html
TreeSAAP can be downloaded from http://genome.byu.edu/treesaap.htm.