
Eric Freeman and Dr. Laura Bridgewater, Microbiology
Collagen is the most abundant protein found in the human body and is responsible for the development of bone and cartilage. The Col11a2 gene is one of the many genes that participate in the formation of collagen. Mutations in this gene can cause a variety of cartilage diseases and disorders such as cleft palate, hearing loss, osteoarthritis, and perhaps even prenatal lethal chondrodysplasia. Arthritis alone is “the leading cause of work-related disability and the leading cause of disability in persons >65 years in the United States”.i The deadly effects of these mutations warrant a more in-depth study of the Col11a2 gene. To determine how the gene is controlled, Dr. Bridgewater’s laboratory performed mutational analysis along the length of the entire gene. Results showed remarkably decreased levels of activity when three locations are mutated; these regions were named ABC, DE, and FG. These areas of decreased activity must be enhancer elements that regulate the transcription of the Col11a2 gene. The research I propose to carry out is that of identifying the transactivating proteins that bind specifically to the FG enhancer element, using electrophoresis, affinity chromatography, and mass spectrometry techniques.
The specific use of the above mentioned techniques in this of experiment was the original idea of colleagues in the lab. The main focus of our research was thus to determine a protocol that would provide us with the desired results, identification of the transactivating proteins involved with the three enhancer elements. I worked with two other students in the lab to establish this protocol using the DE enhancer element. We first used an electrophoretic-mobility shift assay (EMSA) to determine which proteins bind to the DE enhancer under normal conditions. This is done by combining a radioactive copy of either a wild-type or mutant version of the enhancer with nuclear extract that contains the proteins that are present in the cell. This combination allows for various protein-DNA complexes to be formed which when subjected to an EMSA separate into discrete bands. These bands are then visualized by placing x-ray film on top of the gel and developing it. When we carried out this process some of the bands that were present in the wild-type lanes were absent in the mutant version of the gene. This absence of a band indicated the inability of a protein to perform its function and bind to the enhancer in a mutant version because the DNA base sequence to which is normally binds has been changed. Such an inability could contribute to the development of the diseases mentioned in the first paragraph.
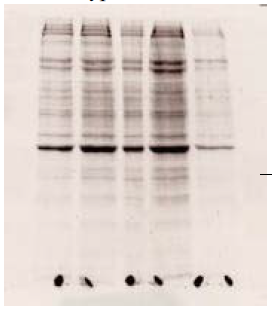

To isolate and identify the missing proteins in the mutant versions of the enhancer we next carried out affinity chromatography using paramagnetic streptavidin beads and biotinylated probes. The beads bound the DNA probes of both the wild-type and mutant sequences. Upon adding nuclear extract the correct proteins (transcription factors) bound to the probes. We added random DNA sequences to remove the non-specifically bound protein. We then separated the desired protein from the probe using SDS and loaded the proteins on a 10% SDS-PAGE stacking gel, which separated the proteins based on size. We stained the gel with SYPRO-Ruby stain and viewed it with ultraviolet light to visualize the bands of protein present. Some of the bands were present in the lanes containing wild-type probe but not in mutant versions (fig.). We excised those bands and subjected the proteins to a trypsin digest, after which we sent the resulting fragments to be analyzed using MS-MS mass spectrometry in order to identify the exact sequence of the protein. The time required to carry out this entire procedure was extensive, but worthwhile. The fragments were analyzed and we were given a large number of possible sequences to compare against a database of protein sequences. We have not as yet found a probable match to the proteins that bind to the DE enhancer element.
After determining that the protocol does provide results I began work on the FG enhancer element. I performed various EMSA’s to determine which mutant versions of the enhancer showed missing bands where expected proteins were not binding. I found missing bands present but unfortunately I was unable to finish the remaining steps of affinity chromatography, SDSPAGE electrophoresis, and mass spectrometry due to unexpected difficulties with lab materials and delays in receiving the mass spectrometer here at Brigham Young University. The rat chondrosarcoma cells (RCS) that provided us with the necessary nuclear extract to perform the experiments became contaminated. The time involved in growing new cells proved more than was available for the remainder of the experiment. I do plan to continue my work with the FG enhancer element throughout this year by using streptavidin/biotin affinity chromatography and mass spectrometry to identify the proteins that are unable to bind to mutant version of the enhancer.
The purpose of our efforts was to determine if the designed protocol would be effective it isolating specific proteins that bind to DNA sequences. Such a method could be published and followed by scientists across the world. Based on this purpose the research carried out provided me with encouraging results showing the effectiveness of the protocol designed. Future experiments will further prove the reliability of the protocol and could result in a future publication. Craig Thulin has helped us with our identification of proteins using mass spectrometry, and our work helped form a new mass spectrometry facility on campus, which will cut down on time required for experiments in the future. Over the course of the year we have also decreased the amount of background (non-specific proteins) in our SDS-gels. My work with the FG enhancer element shows that certain proteins are unable to bind in mutant versions, which leaves room for further experimentation. The results of this work are promising, and I am confident that it will continue to provide important insight into the ways in which gene regulation is carried out and possible solutions to the diseases mentioned at the beginning of this report. Personally, the past year working on this project has increased my understanding of the functions carried out in the cell and has further prepared me for participation in the scientific community.
______________________________________
i JAMA, 272: 346-7 130