Joanna L. Cheatham and Dr. Randy L. Bennett, Zoology
Tribolium castaneum, red flour beetle, is an organism whose development is being studied and compared to that of Drosophila melanogaster. The particular goal of this study was to develop an antibody to Tribolium’s UTX protein, so it’s expression may be monitored during embryonic and larval development.
The fastest way to clone and express a gene is to use a commercial expression plasmid. Scientists use plasmids, which are circular strands of DNA, to introduce a foreign piece of DNA into bacterial cells. Bacteria express genes on plasmids (that is, they make proteins) just as they express genes from their own DNA. This provides a convenient way to make a large amount of the specific protein desired, in this case, UTX. Once the protein is isolated and purified, it can be used to generate antibodies. We chose the “pBAD TOPO TA Cloning kit” from Invitrogen.
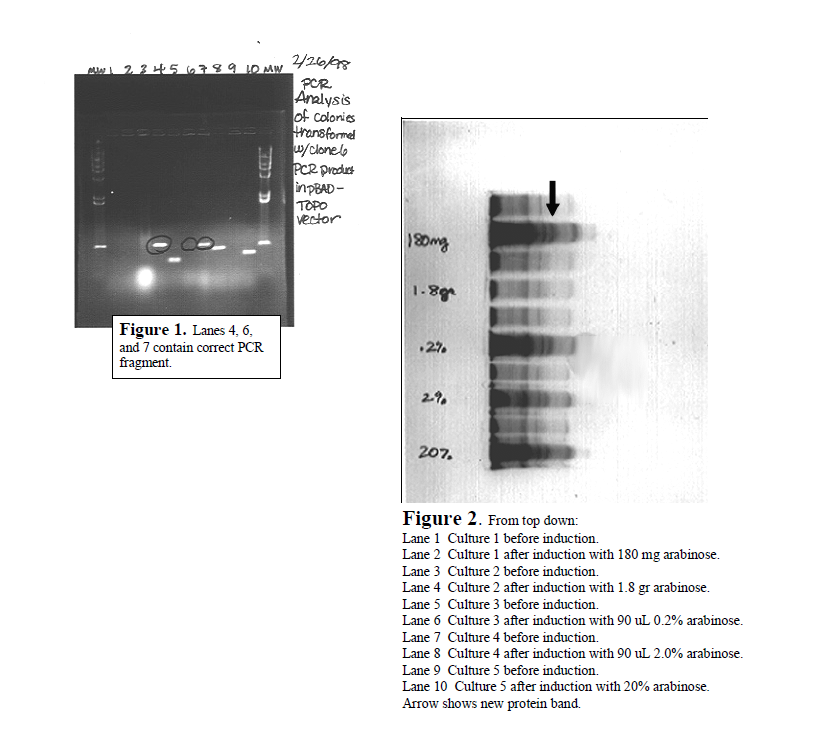
I completed the first major step in the project by cloning the Utx gene fragment into a commercial expression plasmid. First, Dr. Bennett and I designed the primers for PCR. PCR is a technique used to make many copies of DNA from very few copies. I accomplished PCR using an enzyme called Taq DNA polymerase. Taq builds the new DNA fragments, and adds an extra nucleotide, adenine, to the end of each fragment. PCR allowed me to make enough copies of the Utx gene fragment to insert it into the pBAD TOPO vector (plasmid). This vector is what is used to express the UTX protein. Inserting the PCR fragments into the vector was relatively simple. pBAD TOPO is sold with an enzyme attatched to it called topoisomerase. Topoisomerase links the extra adenine from the PCR fragment to the extra thymine on pBAD TOPO. The result is one continuous circle of DNA ready for transformation into bacterial cells. Bacterial cells that are ready to accept plasmids (like pBAD TOPO) are called competent cells, and I accomplished the transformation using competent E. coli cells. The transformation step involved adding the pBAD TOPO vector (with PCR fragments intact) to the cells, and then alternately exposing the cells to hot and cold temperatures. This treatment successfully results in bacterial cells with the plasmid inside. I plated the transformed cells on petri dishes containing nourishing media and ampicillan. The purpose of the ampicillan is to distinguish between plasmids that religated to themselves, and plasmids that incorporated the Utx gene fragment. Only colonies that incorporated the Utx fragment will survive treatment with ampicillan. Ten colonies were picked for PCR analysis, and numbers 4, 6 and 7 looked like they had a fragment the right size to be Utx (see Figure 1). Sequencing these fragments confirmed that the gene was successfully incorporated into colonies 4 and 7 without any mutations.
The next major step was to express and purify the protein. I induced a small volume of cell culture (9 mL) with five different concentrations of arabinose, and found all resulted in induction except the highest concentration. The new protein band, UTX, very clearly showed up on an SDS-PAGE gel (see Figure 2). I then induced a 500 mL culture volume with 0.2% arabinose, spun the cells down into a pellet and lysed them. During protein purification we made the assumption that the protein would be in the pellet of the cell lysate. This assumption was wrong as no protein came off the column from the pellet. The protein is therefore assumed to be in the cell lysate supernatant. 50 Figure 2. From top down: Lane 1 Culture 1 before induction. Lane 2 Culture 1 after induction with 180 mg arabinose. Lane 3 Culture 2 before induction. Lane 4 Culture 2 after induction with 1.8 gr arabinose. Lane 5 Culture 3 before induction. Lane 6 Culture 3 after induction with 90 uL 0.2% arabinose. Lane 7 Culture 4 before induction. Lane 8 Culture 4 after induction with 90 uL 2.0% arabinose. Lane 9 Culture 5 before induction. Lane 10 Culture 5 after induction with 20% arabinose. Arrow shows new protein band. This is where the project stands presently. I had a few delays during the cloning process, such as bad E. coli cells from Invitrogen, that set me back a few weeks. Although I did not obtain the antibody, I finished many critical steps of the process. The gene was cloned into the correct vector and successfully transformed into E. coli. I made glycerol freezes of transformed cells and determined the induction conditions, so doing another growth/induction will be very simple. The cell lysate supernatant is still frozen away and can still be run over the column. The remaining steps in this project are: isolation and purification of the protein, production of antibody, and purification of antibody. This project is well on its way to completion. I graduated from BYU in April and turned the project over to another undergraduate to finish.