Andre Bennin and Dr. Keith A. Crandall, Microbiology and Molecular Biology
Cave microbiology has been of great interest to microbial ecologists in recent years because it has enabled them to assess microbial diversity within an isolated system. An excellent example of subterranean microbial life is found in Lechuguilla Cave Carlsbad Caverns National Park, New Mexico,where the interactions of microbial communities in cave deep subsurface environments have been studied. With respect to the Timpanogos Cave system in American Fork Utah, our study relates to a system that is highly visited (60% of the cave system) contrary to the Lechuguilla cave system where public access is only allowed into 2% of the cave system and tours are kept to a minimum of 10-20 groups per year. Approximately 80,000 people each year introduce mud, hair, lint, and debris into the Timpanogos cave system. The accumulation of these foreign materials is a threat to the natural cave environment. They can change the appearance of cave formations, add alternate energy sources allowing non-native species to invade the cave ecosystem, add impurities to the cave water, and can change the cave’s chemistry. This accumulation of foreign materials threatens the preservation and conservation of the Timpanogos Cave System as a National Monument.
The goal of our research was to document microbial communities in pristine and disturbed areas of the cave. The specific objectives of this project are to: 1) characterize the microbial biodiversity in different areas of the cave; 2) determine if the microbial communities are different in pristine and disturbed sites; and 3) identify unique cave microbial fauna. Comparisons between pristine and disturbed sites were made to see if there were differences in community composition and demonstrate if large numbers of tourists have affected the cave microbial ecosystem.
We collected samples from 10 different sites in the cave system. These samples ranged from water, fungal, fine clay sediments, wood, and mud. Molecular based surveys of 16S ribosomal DNA and phylogenetic analyses were used to determine community composition and relative abundances of bacteria and archaea in the cave. We extracted DNA out of the samples then performed Polymerase Chain Reaction (PCR) on the samples we were able to extract DNA from. The samples that showed bright bands were cloned, and then sequenced after a series of screening test. We completed a phylogenetic analysis of the samples to determine the taxonomic affiliation of each clone. In addition, the ribosomal database project (http://rdp.cme.msu.edu/index.jsp) was used to identify chimeric sequence constructs from the obtained sample sequences. Identified chimeric sequences were removed from our phylogenetic analyses.
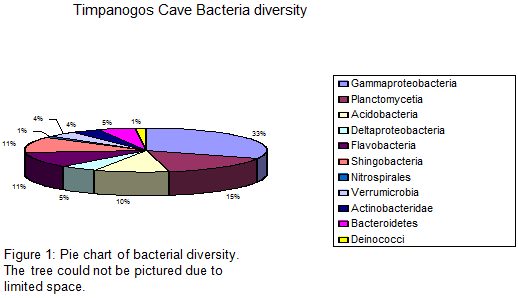
We have sequenced a total of 113 bacteria clones and 39 archaea clones. Five major groups were identified for the obtained bacterial analyses, which include: Gammaproteobacteria, Acidobacteria, Planctomycetia, Sphingobacteria and Flavobacteria (Fig. 1). 55% of the obtained samples from disturbed site TC9R were related to pseudomonas and gammaproteobacteria and the remaining 45% were related to agricultural soil flavobacteria, suggesting a potential impact of microbial communities within this site. 85% of the samples obtained from site TC8R (disturbed) were related to uncultured soil bacteria. The remaining 15% were related to Agricultural soil flavobacteria, suggesting a slight impact of this microbial community from foreign non-native organisms. Samples obtained from site TC7R (disturbed) were closely related to proteobacteria.
All of the obtained archaea samples have come from TC3.2R and T10R, which are pristine sites; we have also obtained clones from TC7R, a disturbed site. Most of the samples showed relatedness to crenarchaeotes and soil crenarchaeotes (Fig. 2). The largest clone archaea group consisted of 70% mesophilic soil crenarchaeotes and front range soil crenarchaeotes. Metal-rich associated archaea represent the remaining 30% of our sampling. Tourist impact will be discussed when more archaea samples are obtained from disturbed sites.
Additional samples were obtained from a new disturbed site on subsequent visits to the cave. Four samples were obtained from this site and were related to environmental proteobacteria Yersinia species.
Bacterial communities have shown to be more diverse than archaea communities in this system. Results have also shown that community composition between sites is different. We obtained archaea sequences from two pristine sites and they have shown similarity in groups; however, we have not obtained archaea sequences for disturbed sites, which prevent us from efficiently comparing the biodiversity between sites for archaea samples. We are still in the process of sequencing more archaea samples in order to make a better comparison of the archaea biodiversity in the cave system.