
Gabriel Proulx and Dr. Keith Crandall, Biology Department
Sequencing genomes still requires considerable equipment and resources. While a full sequencing of all known species is obviously the end goal, analyzing ESTs, short sequences of expressed genes, can suffice in the meantime as an easier and less resource-intensive method to mapping out a species’ expressed genes. ESTs, however, are limited in both length and quality.
While analyzing ESTS from Gossypium raimondii, a variety of cotton, BYU’s Dr. Joshua Udall noticed that many of the DNA reads appeared to display low complexity. This is characterized by long contiguous strings of the same DNA base rather than a more random looking sequence that would be expected. Because of the inexpensive and useful nature of EST sequencing and mapping, and the prohibitive cost of a full genomic sequencing, it is desirable to improve EST datasets by throwing out sequences of low-complexity.
The goal of this project was to create a program for Dr. Udall to use that would not only analyze a sequence’s complexity and throw it out if it contained too many repeating bases, but would do this according to a statistically sound threshold that would be determined by analyzing EST sequences already submitted to the online database Genbank. In order to achieve this goal, I needed to show that the complexities of a species ESTs were normally distributed and that a statistically sound threshold of complexity could be ascertained using a mean complexity and standard deviation.
I downloaded all of Genbank’s EST sequences for several model organisms including, among others, cow, rat, human, and two varieties of cotton. Using scripts written in PERL I analyzed the sequences’ complexities and plotted the results in histograms. This was a formidable task since the sequences for the model species were several gigabytes. I also had to do most data manipulation in PERL since Excel could not handle large enough datasets to accommodate organisms like humans which had 8,000,000 EST sequences.
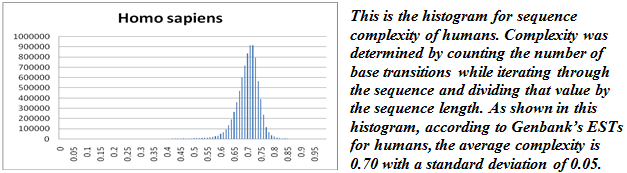
Analyzing the complexities of the model organisms I selected yielded some interesting results. While most of the species I analyzed were normal, a few of the species showed skewed distributions or slight bimodal properties. Gossypium raimondii showed a notable left skew and bimodal properties due to a significant number of low-complexity sequences. Because this was the smallest dataset of those analyzed, it seemed particularly susceptible to bias or bad data, which is what I concluded was the case since it differed markedly from the other graphs. In fact, most of the sequences had come from the same source as the sequences that originally alerted Dr. Udall to this problem. This analysis of complexity highlights the necessity for high quality in lab work, especially when the data will subsequently be submitted to an online database.
Satisfied that my use of statistics was justified for this problem, I moved forward with automating the process of sequence analysis, threshold determination, and sequence sorting. I wrote a program called getcomplex.pl which prompts the user for a genus, species, sample size, and number of blocks to break the sample size into and determines the mean complexity and standard deviation. This data is used in conjunction with my second program, sort.pl, which prompts the user for a mean complexity, standard deviation, the number of standard deviations to use in calculating the threshold, and a file of sequences. Using this data, it sorts the sequences into two new files, those above the threshold and those below. Therefore, when a researcher is analyzing a new set of EST sequences and uses these two scripts with the default values, any sequence with a complexity in the lowest 5 percentile of sequences in Genbank is put into the low-complexity file and should be thrown out or at least scrutinized to guard against using bad data.
In order to extend the usefulness of these scripts, I programmed these scripts to allow for different percentiles, to output histogram distribution, and to dynamically download the data from Genbank each time a query is made. Using different percentiles allows the researcher to specify the level of confidence desired in order to either minimize false positives or false negatives. Outputting histogram data allows the user to analyze the complexity distribution for a species and confirm that it is indeed normally distributed and that there are no abnormalities that would interfere with the threshold. Downloading new data each query allows the scripts to work with every species currently stored in Genbank and to get the most updated sequences.
Both scripts were used on sample data given to me by Dr. Udall. The scripts successfully analyzed the sample data and separated low and high complexity sequences. However, the scripts are limited by the fact that they work on the assumption that the data is normally distributed. It does not work in situations where the complexities of a species’ ESTs are not normally distributed for any reason, such as is the case with Gossypium raimondii where the Genbank data seemed to be biased due to previously submitted low-complexity sequences. The scripts also perpetuate any inherent biases in the Genbank dataset—if the sequences are of a lower complexity, the threshold complexity will be lower and less discriminatory. Finally, the scripts have no real way of telling whether the sequence was in fact bad data or it was merely biologically an actual low-complexity sequence, they can only determine where it stands in relation to Genbank submissions.
Through this project I was first able to successfully analyze the sequence complexity distribution of several model organisms. This projected culminated in the creation of two scripts that allow the user to get a custom sequence complexity threshold for any given species and sort out low-complexity sequences that could hinder a proper analysis. A more complete write-up and the two scripts can be found at http://psoda4.cs.byu.edu/~arkangel/complexity/.