Shaela Avery and Dr. Robert Seegmiller, Physiology and Developmental Biology
Introduction
A recently discovered collagen gene mutation, spondyloepiphiseal dysplasia congenita (sedc), is found on the Col2a1 gene in both mice and humans. The mouse mutation is molecularly similar to the human SEDC mutation, which causes premature osteoarthritis. Studies, such as this one, in mouse models provide a greater understanding of this recently discovered mutation and help in finding ways to prevent osteoarthritis in humans.
The purpose of my study was to assess the amount of knee joint cartilage degradation over time in mice homozygous for the sedc mutation (sedc/sedc) and in mice without the mutation (+/+). I accomplished this by taking the knee joints of mice from different age groups (2, 6, and 9 months), measuring the amount of cartilage within each age group, and comparing the mutant and wild type species to see the difference in cartilage thickness.
Materials and Methods
The animals were euthanized with carbon dioxide, and then I dissected the right knee joint from each mouse. The joints were placed in 4% paraformaldehyde for twenty-four hours to be fixed. Next the knee was decalcified in diluted formic acid for two weeks followed by dehydration in the Tissuematon. The joints were then embedded in paraffin blocks and sectioned at six microns, mounted on slides subbed in aminoalkylsaline, and stained for histological analysis with Hematoxylin and Eosin (H&E) or with Safranin O and Fast Green (SafO & FG).
I examined the prepared slides using SPOT Advanced and Adobe Photoshop CS by taking photos of the tissue at 100X magnification, saving them as jpg images, and opening the images in Adobe Photoshop for analysis. I used the tools in Photoshop to measure total cartilage thickness, thickness of calcified cartilage, and thickness of uncalcified cartilage. During analysis I also looked for differences in H&E stain intensity of the matrix, arrangement of cell nuclei, alignment of chondrocytes, the location of the tidemark (line between calcified and uncalcified cartilage), as well as overall structural differences between the wild-type control and the mutant. SafO & FG revealed further information on the cellular matrix and location of proteoglycans .
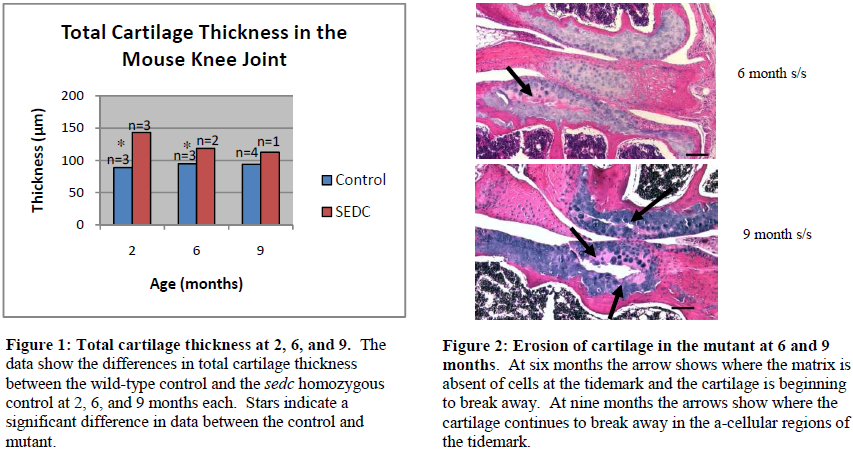
Results and Conclusion
From my histological analysis the sedc homozygous mutant displays significant histological differences in comparison to the wild type control. In the control mouse I saw an even dispersion of cartilage cells within the extracellular matrix as well as columnar alignment of the cells. The mutant, on the other hand, displayed disorganization and clustering of cartilage cells. At two and six months the overall total cartilage thickness in the knee joint of the mutant was significantly greater than that of the control. At nine months, there was no significant difference in the total cartilage thickness of the knee joint between the control and the mutant. In fact, there was a steady decrease in the total cartilage thickness of the mutant over time, whereas the total cartilage thickness of the control remained about the same (see figures below).
Upon further analysis, I was able to determine from my research that the increased cartilage thickness in the sedc homozygous mutant knee joint is due to enlarged lacunae surrounding the cells of the mutant. These enlarged lacunae are proteoglycan rich as indicated by the intense Safranin O staining. The Saf O & FG analysis also indicated a lack of matrix in the knee joint cartilage of the mutant. This could be due to the enlarged lacunae surrounding the mutant cartilage cells. It is possible that some of the proteins necessary for cartilage construction are being trapped within the space around the cells (which explains the enlarged lacunae) and are, therefore, unable to enter the matrix for cartilage construction.
In the control, I expected to see some cartilage degradation as a result of decreased synthesis of extracellular matrix and decreased cartilage construction as the mice aged, but I was uncertain as to how soon cartilage degradation would begin and to what extent this degradation would be in the mutant. At two months, I immediately noted decreased extracellular matrix in the sedc homozygous mutant, which led to premature and extensive degradation in the mutant at six and nine months. Rather than this degradation beginning at the surface of the joint articular cartilage as I had hypothesized, it focused around the tidemark in the mutant where I also noted a lack of cartilage cells at six and nine months. In one of two mutant mice the tissue began to separate at the tidemark at six months. The separation was more prominent in the tibia at six months, but at nine months the separation was greater in the tibia and also seen within the femur.
From my results I conclude that the sedc homozygous mutant mouse is a model for the study of SEDC in humans, because there are significant differences at younger age groups (when SEDC appears in humans). Gaining a better understanding of the enlarged lacunae and cartilage matrix structure within the mutant will help determine whether or not drug therapy is a possible treatment for the sedc mutation, because if the necessary proteins are unable to leave the cell or pericellular space, drug therapy could allow the proteins to enter the matrix for articular cartilage construction.