
Samantha Jensen, Stephen Piccolo, Biology
Precision medicine is a growing movement toward utilizing molecular diagnostics to guide medical decisions. It is particularly useful when applied to cancer treatment, as knowing details about cancer stage, genetic pathology, and tumor type can inform life-saving decisions. Increasingly, physicians may use genetic, proteomic, epigenetic, and expression data to determine treatment strategy and even choose specific chemotherapy drugs1.
Machine learning algorithms can parse large amounts of biological data and find patterns in order to categorize individuals. For example, a computer can now differentiate between acute lymphoblastic and acute myeloid leukemias based only on DNA microarray gene expression data. As the two leukemias have different treatment regimes and pathologies, this information is crucial to maximize efficacy and minimize toxicity of treatment2. After training with gene expression information categorized by tumor type, when presented with new unlabeled data the algorithm could quickly and accurately predict leukemia type. What previously took a hematopathologist’s interpretation of the tumor’s morphology, histochemistry, cytogenetic analysis, and immunophenotyping to distinguish can now be accurately determined by a computer with a single biological sample.
Precision medicine has already saved lives and improved our understanding of cancer pathology3,4. It can be a part of early detection, malignancy prediction, outcomes estimates, and even treatment determination. But it will not reach mainstream adoption until the models of decision making – the patterns computers use to differentiate and predict – are extremely accurate and reliable. And the accuracy of such models is heavily influenced by the type of data used to train them5,6.
Despite the hundreds of millions of dollars currently being invested into generation of data for this purpose, we have little understanding of which genomic data types are most useful in machine learning algorithm design. Researchers use a variety of machine learning methods and whatever biological data they have available to them7,8,9. This project is part of a larger effort to make the selection of biological data for machine learning model development more intentional by comprehensively comparing the performance of different types of molecular data in different cancers with a variety of machine learning algorithms to determine the most accurate combinations.
We used three of the most common machine learning models from the machine learning package sklearn: a multilayer perceptron, a random forest, and a support vector machine (SVM). In three different cancers – urothelial bladder carcinoma (BLCA), kidney renal clear cell carcinoma (KIRC), and lung adenocarcinoma (LUAD) – we used machine learning with six different biological datasets available from the Cancer Genome Atlas (TCGA) to predict different classifications. Each combination of machine learning algorithm, classification scheme, and biological data type was run 10 times to get a more detailed picture of algorithm performance. We used the area under the receiver operating characteristic curve (AUROC) as a measure of how accurately each iteration was able to predict the given class.
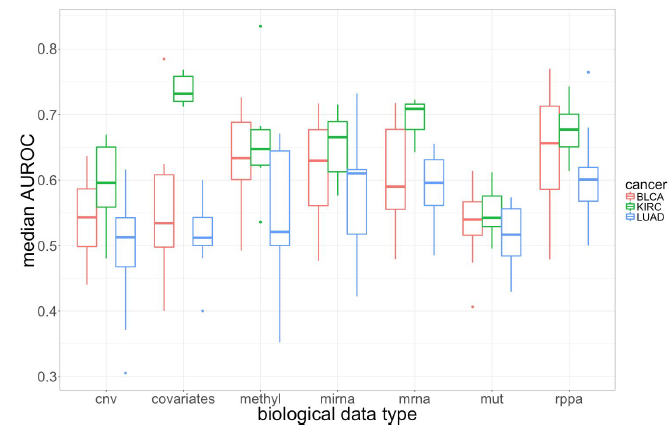
Figure 1 – Median performance of biological data types across all iterations in each cancer. Each cancer had a few different classification schemes (for example, for KIRC we predicted both metastasis and survival), however this is not represented in this graphic. Also not represented is the effect that the machine learning algorithm had on the median AUROC over all 10 iterations. Each biological data type worked best with a different algorithm. For example, copy-number variant (CNV) data was most predictive with a random forest algorithm. These boxplots were created with the median values for all 10 iterations of each combination of cancer, classification scheme, biological data type, and machine learning algorithm.
As you can see in Figure 1, KIRC classifications were more accurate on average across all biological data types. Specifically, it performed exceptionally well with just the covariate information, which is patient demographic information like white blood cell count, gender, and race. This could be because the covariates for the KIRC patients were more directly related to the classification schemes for the cancer, but it also could indicate that KIRC data is more amenable to machine learning approaches.
Most importantly, we found that in general protein expression (rppa) and gene expression (mrna) data were the most predictive and DNA mutation data (mut) the least. None of the three cancers in this initial study have strongly genetic components, so it makes sense that the unique patterns of gene expression and corresponding levels of protein in these patients would most predict survival, metastasis, tumor types, and other classifications.
As we continue to add cancers and algorithms to these initial results, we will be able to make more nuanced predictions about how biological data types will perform in machine learning analyses. The scope of the benchmarking analysis these initial findings are a part of far exceeds any that has been conducted to date and will provide researchers with empirical performance and insights into which combinations of data types and machine learning algorithms provide the most value for informing cancer-treatment decisions.
1 Kourou, Papaloukas, and Fotiadis, “Identification of Differentially Expressed Genes through a Meta-Analysis Approach for Oral Cancer Classification.” 2 Golub et al., “Molecular Classification of Cancer.” 3 Hoesli et al., “Genomic Sequencing and Precision Medicine in Head and Neck Cancers.” 4 Leff and Yang, “Big Data for Precision Medicine.” 5 Yuan et al., “Assessing the Clinical Utility of Cancer Genomic and Proteomic Data across Tumor Types.” 6 Gómez-Rueda et al., “Integration and Comparison of Different Genomic Data for Outcome Prediction in Cancer.” 7 Nahid and Kong, “Involvement of Machine Learning for Breast Cancer Image Classification: A Survey.” 8 Huang et al., “Open Source Machine-Learning Algorithms for the Prediction of Optimal Cancer Drug Therapies.” 9 Kinar et al., “Development and Validation of a Predictive Model for Detection of Colorectal Cancer in Primary Care by Analysis of Complete Blood Counts: A Binational Retrospective Study.”