
Meganne Ferrel and Dr. John S. K. Kauwe, Biology
Introduction
Alzheimer’s Disease (AD) is identified as a proteopathic disease that results from an extensive accumulation of amyloid plaques and neurofibrillary tangles in the brain. Several researchers have discovered that cerebrospinal fluid (CSF) contains biomarkers for AD such as Amyloid-beta and tau. Neuritic plaques in AD are surrounded by activated microglia and astrocytes which can initiate complement and inflammation in the presence of amyloid-beta. Macrophage inflammatory protein chemokine (C-C motif) ligand 4 (CCL4) is a biomarker found in CSF. A high level of expression is associated with risk of developing AD. CCL4 is expressed in a subpopulation of reactive astrocytes and microglia. Genome-wide Association study (GWAS) has identified a possible association between an atypical C-C motif chemokine receptor-like 2 (CCRL2) and CCL4. Mutation CCRL2-V180M shows to be statistically associated with decreased CSF CCL4 protein levels. However, due to the atypical nature of CCRL2, it has previously been suggested on a molecular level that CCRL2 and CCL4 do not interact. We hypothesized that the mutant CCRL2-V180M has altered binding affinity and functional response to CCL4.
Methodology
We constructed plasmids with the chemokine receptor-like CCRL2-WT wild type (WT) and a plasmid carrying CCRL2-V180M. These plasmids were transfected into Chinese hamster ovary (CHOK1) mammalian cells; a separate plasmid with GFP was also introduced to visualize transfection rate. Cells were then selected using G418 Neomycin. Cells were cultivated using Ham’s F-12K medium. Precise transfection rate was determined using anti-human CCRL2 antibody. A competitive binding assay utilizing flow cytometry was performed between receptor-blocking antibody and the chemokine ligand CCL4 to determine binding affinity and interaction between CCRL2 and CCL4.
Results

Discussion
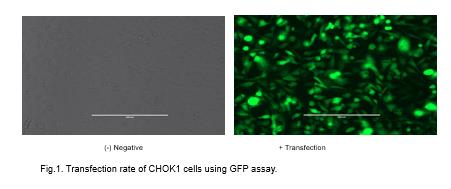
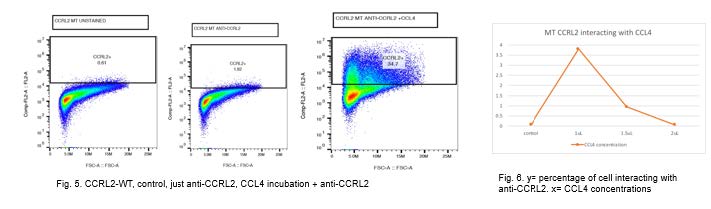
Plasmid transfection rate resulted in over 92% transfection (Figure 1). By adding 1ng of CCL4, no change of interaction between anti-CCRL2 and CCRL2 receptor as compared with the control was visualized. Upon the addition of more CCL4, there was an inhibition of interaction between the anti-CCRL2 and CCRL2 receptor (Figures 3 and 4). On the other hand, addition of 1 ng of CCL4 to the mutant receptor demonstrates that there was an increase of binding interaction between anti-CCRL2 and the CCRL2 receptor. Higher concentration of CCL4 down regulates the initial interaction between the anti-CCRL2 and mutant receptor.
Conclusion
Preliminary results have determined that CCRL2 and CCL4 may have a binding interaction. In addition, we observed that CCRL2=WT and CCRL2-V180M have different binding affinity response in the presence of CCL4. These results show a potential pathway to lower levels of CCL4 and diminish inflammation, resulting in decreased AD progression. This could lead to novel therapeutic approaches for Alzheimer’s Disease.