
Gage Black and John S. K. Kauwe, Biology
Introduction
Alzheimer’s Disease (AD) is the most common form of dementia and affects millions of people worldwide. It is the 6th leading cause of death in the United states and the only disease in the top 10 causes of death that does not have a disease altering treatment. Due to the complex nature of the disease, it is difficult to confirm whether a patient has AD or a different form of dementia. Misdiagnosis of AD and other dementias could be leading to the use of inaccurate data in genetic studies specific to AD. In 2014, Beecham et al. collected data from 5,660 AD cases and controls confirmed through neuropathological diagnoses. Using this data, we have assessed the genetic heterogeneity between neuropathologically confirmed AD subjects and clinically diagnosed AD subjects by comparing their proportion of total phenotypic variance. This comparison gives insight into the efficacy of using data collected from clinically diagnosed individuals in AD research.
Methods
We obtained a single nucleotide polymorphism (SNP) dataset of clinically diagnosed individuals from the Alzheimer’s Disease Genetics Consortium (ADGC) comprising of 30 separate studies that were combined and filtered by Boehme et al. Duplicate SNPs were removed and a minor allele frequency threshold of 1% was set to increase the quality of our dataset. Any sample missing a covariate for age, sex, or status was also removed. Individuals with neuritic plaque and classified with a Braak Stage 3-6 were declared as AD positive. All other individuals were considered AD negative. Genome-wide Complex Trait Analysis (GCTA) allowed us to create a genetic relationship matrix (GRM) for each chromosome within both datasets of clinically diagnosed individuals and neuropathologically confirmed individuals. A restricted maximum likelihood (REML) analysis was performed using the GRMs generated by each dataset to estimate the variance explained by all 8,712,879 SNPs. Results were considered statistically significant if the p-value provided by the REML analysis was less than 0.05. A T-test was used to compare genetic variance explained at the overall and chromosome specific levels between the neuropathological and clinically diagnosed subjects.
Results
The estimated proportion of the total phenotypic variance explained by all SNPs was not significantly different (p-value 0.725) between the neuropathologically confirmed dataset (50.13% n = 3084), and the clinically diagnosed dataset (55.69% n= 8801) (Figure 1). Chromosome 19 was the only location that showed a statistically significant difference in genetic variance explained between the two datasets (p-value 0.0001) (Figure 2).
Discussion
Our analysis suggests there is not a significant difference between clinical and neuropathological diagnosis in Alzheimer’s Disease. Overall, this suggests that clinical diagnosis as conducted at the sites involved in the ADGC study are quite reliable in discerning AD from other dementia in research subjects. Chromosome 19 does explain a significantly lower proportion of total phenotypic variance in neuropathologically confirmed individuals relative to clinically diagnosed subjects. We find this interesting because this is the location of APOE, a gene that can influence your risk for late onset Alzheimer’s Disease. Further analysis of this chromosome will allow us to gain a better understanding of what might be driving the differences being observed.
Conclusion
The significant difference in chromosome 19 implies that clinically diagnosed subjects may include those with other dementia or increased risk of sporadic AD that is driven specifically by variants in chr 19 (perhaps APOE). However, there is not a significant difference found in the total phenotypic variance explained by all SNPs between neuropathologically confirmed and clinically diagnosed individuals.

Figure 1 – Phenotypic variance explained by all SNPs by chromosome. Error bars correspond to standard error.
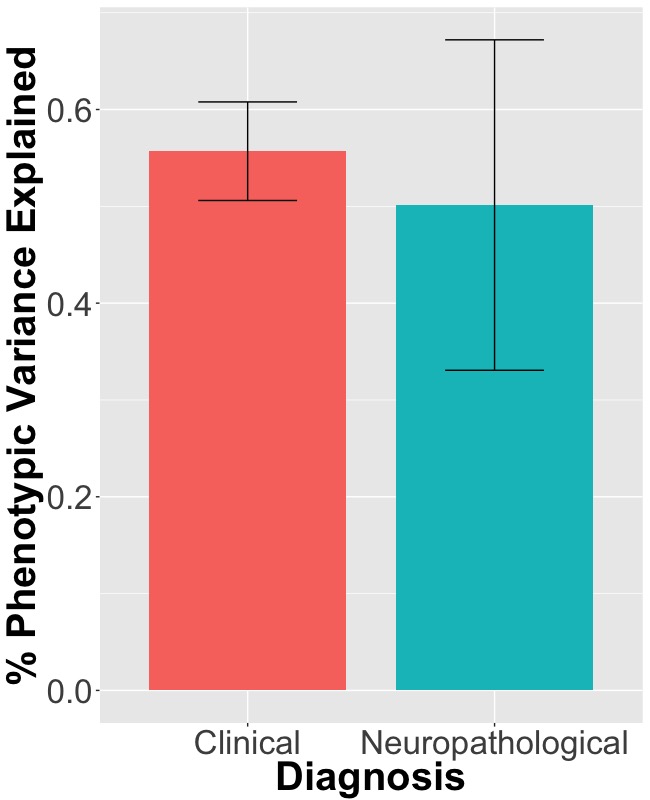
Figure 2 – The overall proportion of phenotypic variance explained by all SNPs. Error bars correspond to standard error.

Table 1 – Demographic information for individuals in the analysis dataset and summary of phenotypic variance measurements.