
Amanda Hobson and Jeffrey Tessem, Nutrition, Dietetics, and Food Science
Introduction
Type 1 and Type 2 diabetes are classified as a decrease in functional ß-cell mass, which results in impaired blood glucose regulation. Functional ß-cell mass is defined as the glucose stimulated insulin secretion rate multiplied by the total cellular mass which is determined by the proliferation and cell death rates. Though ß-cell mass proliferation rates generally decrease by adolescence, obesity and pregnancy have been shown to be times of significant ß-cell proliferation (1). This implies that the inherent molecular pathways necessary for ß-cell proliferation are present, but highly regulated. Discovering the molecular pathway of ß-cell proliferation could lead to diabetes treatment options including increasing the endogenous ß-cell population, or producing sufficient ß-cells ex vivo for islet transplantation.
In the ß-cell proliferation pathway, Nkx6.1 has been shown to drive ß-cell proliferation, protect against apoptosis, and enhance glucose stimulated insulin secretion (2). Nkx6.1 has also been proven to induce Nr4a1 and Nr4a3 nuclear receptors which drive ß-cell proliferation (3). We performed expression analysis of genes upregluated by Nr4a1 and Nr4a3 but not Nkx6.1 to discover what factors regulated by Nr4a1 and Nr4a3 are sufficient to induce ß-cell proliferation. We found that the unique neuron specific cyclin family member CDK5R1 was upregulated by Nr4a1 and Nr4a3, but not Nkx6.1. We also found through Edu incorporation assays that CDK5R1 induces ß-cell proliferation. Our lab has also shown that increased levels of CDK5R1 maintain insulin secretion rate, effectively maintaining the mature ß-cell phenotype. Several studies have demonstrated that genes that induce ß-cell proliferation rates also decrease insulin secretion rates, or increase apoptosis rates. Before CDK5R1 can be considered as a viable treatment option for increasing functional ß-cell mas, we must determine the effects of CDK5R1 on ß-cell apoptosis.
Methodology
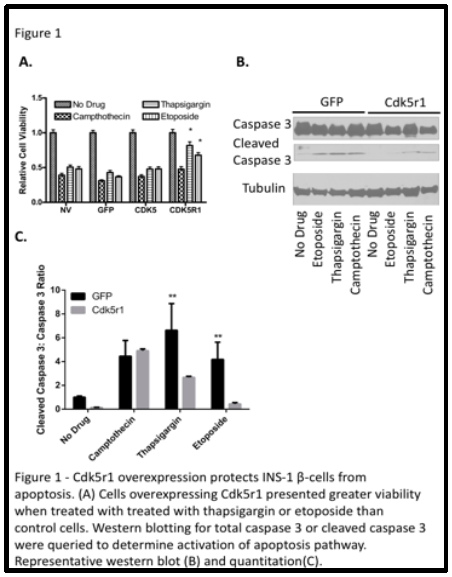
For the experiments conducted testing the effect of CDK5R1on ß-cell apoptosis, we used the INS-1 832-3 rat insulinoma ß-cell cell line. We transfected the INS-1 832-3 ß-cell line with ß-gal (viral control), Nkx6.1 (positive control), CDK5R1 (experimental factor), or untreated (negative control) for four hours. Forty-eight hours after transfection, we exposed the cells to the apoptotic drugs etoposide (0.9μM), campthothecin (2.0μM), and thapsigargin (0.32mM). After being exposed to the apoptotic drugs for 18 hours we did cell counts to determine ß-cell survival. A total of nine replicates were completed.
A second set of experiments, which included western blotting, were conducted to confirm the rate of apoptosis in cells that were transfected with CDK5R1. We measured the amount of cleaved caspase 3 protein which is indicative of cell death. The INS-1 832-3 cells were transfected with virus and treated with apoptotic drugs following the same time line that was used for cell counts. The ratio of cleaved caspase 3 to total caspase 3 was measured and quantified.
Results
Our results showed apoptotic protection in ß-cells overexpressing CDK5R1 that were exposed to etoposide and thapsigargin. This means that CDK5R1 protects ß-cells from Ca2+ induced ER stress, and topoisomerase II inhibitor induced apoptosis. However, we did not see any protective effect of CDK5R1 on ß-cells treated with campthothecin, meaning that CDK5R1 overexpression does not protect against topoisomerase I inhibitor induced apoptosis.
We also saw lower levels of cleaved caspase 3 to caspase 3 protein ratio in ßcells overexpressing CDK5R1 that were exposed to thapsigargin and etoposide. Higher levels of cleaved caspase 3 to caspase 3 protein levels indicates an increase in the apoptotic pathways. We did not see a decreased ratio in ß-cells exposed to campthothecin which is consistent with the cell viability data collected. Overall, our data shows that overexpression of CDK5R1 protects ßcells against selected apoptotic pathways. The results from these experiments were published in the Journal of Diabetes Research in September 2015.
Discussion and Conclusion
The results from our cell viability assays and western blots prove our hypothesis that overexpression of CDK5R1 protects ß-cells from undergoing apoptosis when treated with etoposide and thapsigargin. In addition to our previous research that has shown CDK5R1 to drive proliferation, and maintain glucose stimulated insulin secretion, we now know its effect in protection against apoptosis. Our data has significant implications for the potential use of CDK5R1 as a therapeutic entry point for increasing ß-cell mass ex vivo for transplantation or for the expansion of endogenous ß-cell mass in diabetic patients.
Scholarly Sources
- A. E. Butler et al., Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia 53, 2167 (Oct, 2010).
- J. C. Schisler et al., Stimulation of human and rat islet beta-cell proliferation with retention of function by the homeodomain transcription factor Nkx6.1. Molecular and cellular biology 28, 3465 (May, 2008).
- J. S. Tessem et al., Nkx6.1 regulates islet beta-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proceedings of the National Academy of Sciences of the United States of America 111, 5242 (Apr 8, 2014).