
Taylor Southwick and Dr. Keith Crandall, Department of Biology
Main text
The classification of organisms into different taxa is a central issue in biology. If similar organisms are grouped together, then comparative analyses can be used from the genome to the systems level to understand how they work, both biologically and behaviorally. There are two ways to perform this classification: morphologically and phylogenetically. Morphological demarcations are based on visual differences in anatomical structure and physiological function. Phylogenetic classification is based upon the evolutionary relationship among the individuals in the population quantified by the similarity or dissimilarity of their DNA strands.
Phylogenetic analysis of relationships has become the method of choice as it is more quantifiable than studying morphological changes. If a full genome were available for each organism in the world, this would be easy; however, such genome wide studies are currently too expensive, tedious and cumbersome. Thus small sections of homologous DNA have been used to discriminate among organisms. The main gene used for this purpose is the COI gene, which is found in most, if not all, organisms. This gene was proposed as a simple way to classify organisms by Paul Hebert in 2003. Since then this technique of organizing organisms has been known as barcoding1.
The gene being used, COI, is a 648-bp region of DNA found within mitochondrial genomes, which most eukaryotic organisms contain. This is mostly used with animals and most eukaryotes, except for flowering plants as the COI in their mitochondria does not evolve as quickly as in other organisms. Criticism has arisen in the past that COI is good for identification, but not for classification2. This comes from several factors, but most notably includes the argument that there is not enough diversity in COI for classification, especially in recently diverged organisms, and that mitochondria are only inherited maternally. Thus a number of factors can skew the genetic data transferred. This project attempted to show by estimated phylogenetic relationships among species of crab that this hypothesis is true. Specifically it demonstrated the COI gene by itself will yield a statistically different topological structure of relationship than a combination of multiple genes from the organisms.
To begin, around two hundred individual crabs were sequenced for five genes. These include: 12S, 16S, 28S, COI, COII. These are a mixture of mitochondrial and ribosomal DNA, which are found in many species and have been used for identification purposes or classification. Once all the sequences were obtained and the quality was ensured, it was necessary to align thesequences. Alignment will indicate where there are gaps in the sequences caused by either insertions or deletions at some point. When sequences are aligned they allow the researcher a view of the similarity or dissimilarity of a sequence is. This is necessary to estimate the relationships among them. This alignment was carried out by MAFFT, one of the better multiple sequence alignment algorithms available.
Once the aligned sequences were acquired a tree could be estimated that would graphically show the genetic relationship among the sequences. This was done with two algorithms: maximum likelihood with RAxML and Kimura-2 parameter (K-2) with PAUP3. Maximum likelihood tends to create a better estimation, but the K-2 algorithm is frequently used, especially for estimating relationships among barcoded organisms. Fig. 1 shows a tree generated by maximum likelihood when all five genes were used as input, while Fig. 2 shows a tree produced with the same method when only COI is used as input.
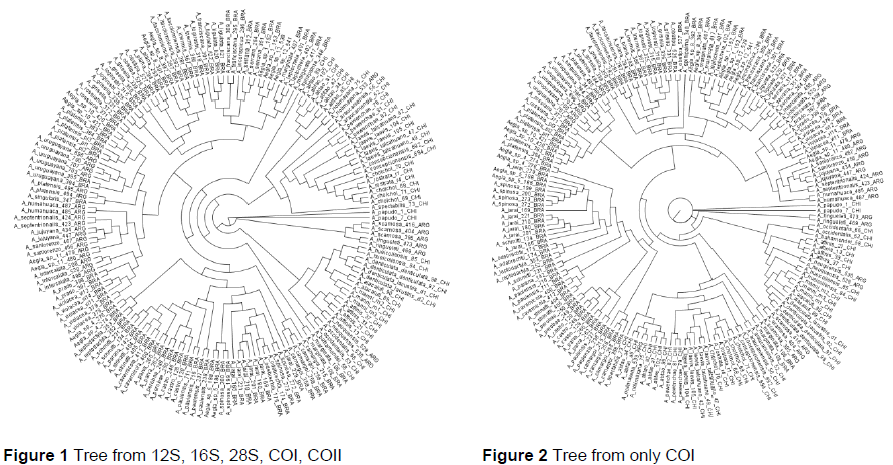
When a statistical test was performed on the resulting trees with SHTest within RAxML, it was found that all of the trees created with just COI differed significantly from the trees created by using multiple genes. Thus, COI itself is not unique enough to fully describe the phylogenetic relationship among organisms and should not be used for classification purposes.
References
- Hebert, Paul D N, Alina Cywinska, Shelley L Ball, and Jeremy R deWaard. “Biological identifications through DNA barcodes.” Proceedings of the Royal Society of London. Series B: Biological Sciences 270 (2003): 313-321.
- Miller, Scott E. 2007. DNA barcoding and the renaissance of taxonomy. Proceedings of the National Academy of Sciences of the United States of America 104, no. 12 (March): 4775-6.
- Stamatakis, Alexandros. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics (Oxford, England) 22, no. 21 (November): 2688-90. doi:10.1093/bioinformatics/btl446. http://www.ncbi.nlm.nih.gov/pubmed/16928733.