
John Daniel Clinger and Dr. Daniel L. Simmons, Chemistry and Biochemistry
Recent advances have greatly increased the utility of bioluminescence as a qualitative and quantitative tool in scientific research. The genes encoding fluorescent proteins in several organisms have been identified, cloned, modified, and inserted into several different vectors; thus allowing them to be used in a variety of diagnostic techniques. One such protein, isolated from aequorea victoria, is green fluorescent protein (GFP). Since GFP is encoded by a small gene, 720 base pairs, and fluoresces brightly when irradiated by ultraviolet light; it was seen as good candidate for the study of a novel pre-mRNA splicing mechanism.
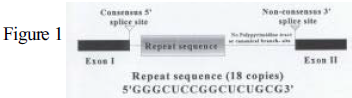
Research conducted in the laboratory of Dr. Daniel L. Simmons led to the discovery of cyclooxygenase-2 (COX-2) in 1991. COX-2 is an inducible isoenzyme that together with its constitutive form, COX-1, catalyzes the rate-limiting step in the conversion of arachidonic acid to prostaglandin G2 (PGG2) and prostaglandin H2 (PGH2). The primary transcript of COX-2 must undergo pre-mRNA splicing to remove intronic sequences, thus allowing translation of the functional protein. It has been found that intron-1 of COX-2 in chicken is only removed when pre-mRNA splicing is stimulated through mitogen-inducible signal transduction pathways. This novel intron-1 of chicken COX-2 is flanked by neither the GT-AG of major class introns nor the AT-AC of minor class introns, but rather GT-CT. Intron-1 also lacks the polypyrimidine tract and internal branch-site present in typical introns; but contains 18 consecutive copies of a 16- nucleotide sequence (Fig.1). These unique characteristics would indicate that intron-1 of COX-2 in chickens is spliced through a previously unknown pathway that depends on mitogens to induce splicing fidelity at the 3′ splice site.¹
Previous research had shown that the region of chicken COX-2 that defines the 3′ splice site of intron-1 is not found in exon-1, the intron itself, or the following 329 bases corresponding to exons 2-4. Our research since these findings has been aimed at determining which elements in exons 5-10 are responsible for the definition of the 3′ splice site in chicken COX-2. Recently we found through the use of deletion constructs that a cis-acting element that defines selection of the unique 3′ splice site is found at nucleotides 1378-1841 of the chicken COX-2 mRNA. Weaker elements that modulate selection of the 3′ splice site were also found at nucleotides 2543-3135.
To follow up the discovery of these cis-acting elements, a construct in which intron-1 of luciferase was replaced by intron-1 of chicken COX-2 had 463 nucleotides, corresponding to nucleotides 1378-1841 of chicken COX-2, placed in the 3′ untranslated region (UTR). Subsequent transfection of this construct into chicken embryo fibroblasts (CEF) and luminescence assays revealed that although the 463 nucleotides were necessary for correct splicing to the 3′ CU site, it was not sufficient to ensure splicing fidelity. This left us with the question of whether the three nucleotides (CCA) just 5′ of intron-1 in chicken COX-2 were sufficient for splicing to the correct 3′ splice site. GFP was selected for these studies.
The 5’ three nucleotide sequence (CCA) was identified at several points along the DNA encoding GFP in the pEGFP vector from Clontech Laboratories, Inc. (Palo Alto, CA). Three different sites were selected as insertion points for chicken COX-2 intron-1. Later the 463 nucleotide cis-acting element would also be placed in the 3′ UTR of each of these constructs. The production of functional GFP in the CEF would be indicative of correct splicing to the 3′ splice site. Specific oligonucleotide primers that would selectively amplify, via polymerase chain reaction (PCR), the flanking fragments of GFP for each of the three constructs and chicken COX-2 intron-1 were prepared. Each 5′ GFP fragment and intron-1 could then be linked using T4 DNA ligase. This ligation reaction would then be probed by PCR using a sense strand primer corresponding to the 5′ GFP fragment and an anti-sense strand primer corresponding to intron-1.The correctly linked and selectively amplified fragment would then be gel purified and linked with the 3′ GFP fragment. This ligation reaction would then be probed with the sense primer for the 5’ GFP fragment and the anti-sense primer fragment for the 3′ fragment. Thus the entire correctly oriented construct could be amplified and inserted into a vector for experimentation.
Upon execution, the GFP fragments flanking the intron-1 insertion points were easily amplified. Intron-1 of chicken COX-2 proved much more difficult to amplify. The amplicons yielded by PCR were smears in which the band corresponding to intron-1 was hardly discernible. Several adjustments were made to the PCR reaction conditions, reagent concentrations, template DNA, oligonucleotide primers, and thermocycler parameters. When optimization attempts seemed futile it was ultimately decided that the guanine- and cytosine-rich 16-nucleotide consecutive repeats of intron-1 did not allow for the clean amplifications necessary for successful ligation reactions and subsequent amplifications.
In order to bypass the problems caused by the repeated sequence in intron-1, the vector containing the entire chicken COX-2 gene was digested with the restriction endonuclease BssHII. Digestion excised the repeated sequence, leaving 95 bases of intron-1, and created a linearized vector that was gel purified and electroeluted. The vector was recircularized using T4 DNA ligase and transformed into supercompetent cells. Clones were cultured, extracted, and confirmed by restriction digest. The recircularized vector was used as DNA template for PCR using oligonucleotide primers specific to the 5′ end of intron-1 and a region 307 nucleotides 3′ of the intron-1 splice acceptor site. This 402 base pair fragment was then linked with the 5′ GFP fragment. The resulting ligation was probed by PCR using a sense strand primer corresponding to the 5′ GFP fragment and an anti-sense strand primer corresponding to intron-1. This linked fragment was then gel purified and digested with BssHII to confirm the presence of intron-1.
Although the 3’ GFP fragment must still be successfully linked and the repeated sequences inserted to complete the construct, I am confident this experiment will provide valuable future information on the nature of this unique intron.
References
- Madsen, M.L. (1997). Mechanistic Studies on the Signal Transduction Regulated Excision of Intron-1 for the Chicken Prostaglandin G/H Synthase-2 Gene During premRNA Splicing. Dissertation, 109-122.