
Alexander Parent and Dr. Steven Fleming, Chemistry and Biochemistry
Organic chemistry centers around the stereo- and regioselective formation of C–C bonds. Although [4+2] and [2+2] cycloadditions present the opportunity for single-step creation of multiple stereocenters and two C–C bonds, the products resulting from intermolecular cycloadditions can be hard to predict and yields of any single isomer can be low. These obstacles can be overcome by the use of a removable tether, which induces regio- and stereo-control through the forced intramolecular reaction. The silyl acetal-tethered [2+2] photocycloaddition has been shown to produce high yields of a single product when both of the reacting alkenes contain extended -systems. This phenomenon is considered as being due to either -stacking between the alkene groups or radical stabilization.
Because of the electron donating nature of the linking oxygen and lack of an extended -system it is proposed that a silyl enol ether moiety would have the ability to radical-stabilize without participating significantly in - interactions. Therefore a cinnamyloxysilane containing an enol ether group should allow for analysis of the effects of -stacking on the silyl-tethered photocycloaddition that is consistent with previous silyl-tethered irradiations. Depending on: 1) whether cycloaddition occurs and 2) the regio- and stereochemistry of the photoproducts formed, inferences can be made concerning the concerted or step-wise nature of the [2+2] photocycloaddition.
After synthesis and purification, the diisopropylcinnamyloxysilyl enol ether of cyclohexanone was irradiated using a quartz jacket and well photolysis set up. The separation and isolation of the various photoproducts was accomplished using various forms of chromatography. Identification of the major photoproducts was accomplished primarily using various forms of nuclear magnetic resonance spectroscopy, including 1H NMR, 13C NMR, COSY, HETCOR, and NOE.

Our initial hypothesis maintained that if the previously-studied silyl-tethered 2+2 cycloadditions proceeded through a concerted mechanism, -stacking was a necessary energy-lowering factor for these reactions. Conversely, if a diradical-mediated mechanism was involved in the photocycloadditions, radical stabilization would prove adequate to produce cyclobutane photoproducts. Consequently, because the silyl enol ether 37 lacked the conjugated -system necessary for -stacking it was expected that if its photolysis produced any cycloaddition products, they would have the regiochemistry shown in 41, forming from the most stable diradical intermediates. The formation of 41 or its stereoisomers would strongly support the presence of the diradical mechanism and marginalize the energy-lowering effects of -stacking in the previously reported silyl-tethered systems.
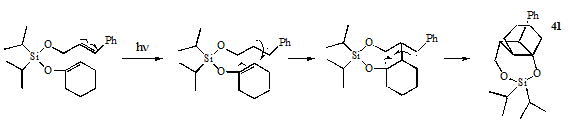
It is important to note that instead of the regiochemistry predicted by the step-wise mechanism, 38 and 39 contain a 6-member siloxane ring instead of the 7-member siloxane ring of 41. This indicates that the cycloaddition does not proceed through the most stabilized diradical, but likely proceeds by a concerted mechanism advancing from the lowest-energy conformations of the starting material, as indicated by formation of the 6- instead of 7-member ring. The two stereoisomers can be explained as arising from the two possible orientations of the cyclohexenyloxy group to the cinnamyl group which leads to the 6-member siloxane ring as shown below.
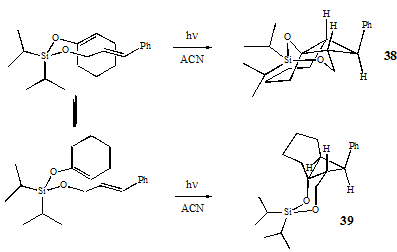
The surprising reactivity of the diisopropylcinnamyloxysilyl enol ether of cyclohexanone can be attributed to the oxygen of the enol ether. Although it first appears that there is only a minimal -system available on the enol ether to undergo - interactions with the styrenyl group, it must be remembered that the lone pair on the enol ether oxygen is in conjugation with the -bond of the alkene. This conjugation not only creates an effective three-atom -system, but one that is also electron rich due to the oxygen lone pair (four -e- shared over three atoms). The increased -stacking ability of the electron-rich three-atom system activates the system towards the concerted cycloaddition, effectively overcoming any favor for the radical-mediated one. Therefore, although the role of radical stabilization in the silyl-tethered [2+2] photocycloaddition has not been elucidated, it is clear that a three-atom -system, specifically an electron-rich one, will undergo sufficient -stacking to activate the tethered, singlet [2+2] system towards photocycloaddition.