
Spencer Jones and Dr. Steven Castle
Abstract
Hasubanonine is a tetracyclic alkaloid with potential analgesic activity. The total synthesis of this alkaloid has been undertaken in order to demonstrate the applicability of the methodology developed for the synthesis of acutumine and to assay the potential medicinal activity of the compound. The key steps in the synthesis include the ruthenium catalyzed ring closing metathesis, anionic oxy-Cope rearrangement, and intramolecular Michael addition. Future work includes the completion of the racemic synthesis of hasubanonine as well as the investigation into the asymmetric allylation required for the enantioselective synthesis of hasubanonine.
Introduction
The synthesis of compounds containing multiple contiguous quaternary sterocenters is a difficult challenge for the synthetic organic chemist. The challenge arises from the difficulty of enantioselectively constructing the centers in a sterically hindered environment. Unfortunately, there are no general solutions to this problem.1
In order to synthesize the natural product acutumine, our research group developed a methodology that enables the construction of vicinal quaternary stereocenters in a sterically hindered environment.2 The methodology developed specifically for the synthesis of acutumine is directly applicable in the total sythesis of hasubanonine and its analogs due to structural homology.3 The core dimethoxyenone structure of acutumine and hasubanonine are nearly identical as can be seen in Figure 1.
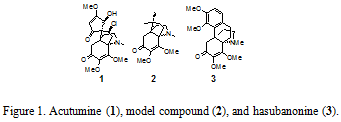
Hasubanonine is a tetracyclic alkaloid with two adjacent quaternary stereocenters, one of which is an all-carbon stereocenter. Because hasubanonine contains one less contiguous stereocenter than acutumine, it is an ideal target to demonstrate the methodology developed for the synthesis of acutimine.
The unnatural enantiomer of hasubanonine is structurally similar to the potent analgesic morphine. Due to this similarity, Shultz et al. theorized that the unnatural enantiomer of hasubanonine could likewise possess a degree of analgesic properties.4
This medicinal activity has not, to date, been tested. Thus, in order to demonstrate of our methodology and test the possible medicinal activity, our research group set out to enantioselectively synthesize hasubanonine.
Methods and Results
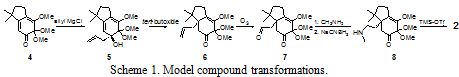
A simplified model 2 of acutumine was synthesized in 2003–2004 by our research group. The final steps in the synthesis of hasubanonine involve the same transformations used to synthesize 2. The key steps of the transformations (shown on the model compound) are as follows (outlined in Scheme 1). Allyl Grignard addition to masked ortho benzoquinone 4 yields tertiary alcohol 5. Deprotonation of 5 promotes an anionic oxy-Cope rearrangement to yield the more thermodynamically stable ketone 6. Selective ozonolysis of the terminal olefin of 6 gives aldehyde 7. The aldehyde of 7 is then reductively aminated with methylamine to give the secondary amine 8. The final cyclization of the amine is facilitated by the Lewis acid TMS-triflate to give the model compound 2.
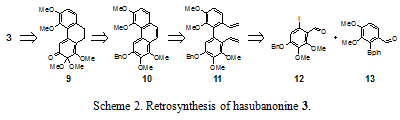
The retrosynthesis of hasubanonine is shown in Scheme 2. Simplification of 3 in accordance with the chemistry described above affords compound 9. Retro-oxidation followed by functional group interconversion provides phenanthrene 10. Again in the reverse direction, ring closing metathesis, functional group interconversion, and palladium-catalyzed Suzuki coupling reveals the two aldehydes 12 and 13
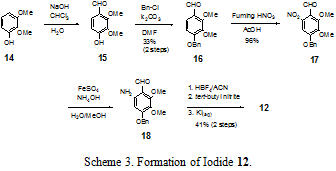
In the forward direction (Scheme 3), the synthesis of 12 begins with the formylation of dimethoxyphenol 14 with chloroform and sodium hydroxide in water to give aldehyde 15 via dichlorocarbene insertion into the carbon-hydrogen bond. Partially purified 15 is benzylated with benzyl chloride to give 16 in 33% yield over two steps. The low yield of the two steps is a result of the carbene insertion into the carbon-hydrogen bond sterically favoring the position ortho to the hydroxyl group, rather than ortho to the methoxy group, as is needed for our synthesis. Aldehyde 16 is nitrated with fuming nitric acid in acetic acid to afford nitrobenzene 17 in 96% yield. Examination of the electronics of the benzaldehyde would suggest that the nitration would proceed at the most electron-rich position ortho to the benzyloxy group. In fact, this was the substitution pattern seen when electrophilic iodine was reacted with the aromatic moiety directly. However, we postulate that in this case the substitution occurs ortho to the aldehyde functionality due to coordination of the nitronium ion with the electrons on the carbonyl oxygen. Reduction of the nitro functionality is accomplished with iron (II) sulfate and ammonium hydroxide in methanol/water to provide amine 18 which is used crude in the subsequent steps. An NMR analysis of the crude reaction mixture shows that it is nearly completely the amine. However, column chromatography of the amine failed to produce yields higher than 30%; for this reason the amine was used crude in the subsequent step. Crude amine 18 is converted into a diazonium salt with fluoroboric acid and tert-butyl nitrite and then immediately quenched with aqueous KI to generate aromatic iodide 12 in 41% from 17. Standard procedures for the synthesis of diazonium salts were investigated for the synthesis of the diazonium salt intermediate from 18. As these all failed to produce any detectable quantity of the iodide, a less standard procedure involving fluoroboric acid and tert-butylnitrite was investigated and found to work satisfactorily.5
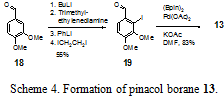
The synthesis of 13 (Scheme 4) involves the directed ortho-metalation of 18 with PhLi followed by quenching with 1,2-diiodoethane to provide iodide 19 in 55% yield. Neither changing the organolithium reagent nor the electrophilic iodine source succeeded in increasing the yield above 55%. Iodide 19 is coupled to bis-pinacolatodiboron in a Suzuki-type coupling with catalytic palladium acetate to afford 13 in 83% yield.
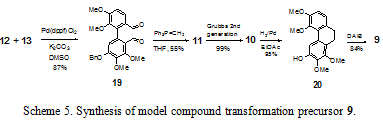
Palladium-catalyzed Suzuki cross coupling of 12 and 13 yields 19 as a mixture of two atropisomers due to hindered rotation about the biaryl axis in 87% yield (Scheme 5). Pd(dppf)Cl2 was found to be the ideal catalytic palladium source for the coupling. The other common ligands for palladium such as triphenylphosphine and tri¬ortho-tolylphosphine yielded a complex mixture of products or reduction of the aromatic iodide to the corresponding hydrogen-substituted aromatic ring. Dialdehyde 19 is converted to diene 11 via the Wittig reaction in 55% yield. Iuliano et al. similarly reported low yields of the Wittig reaction of 2 2’diformylbiphenyl compounds as we observed.6 Potentially, other means of olefination would not be affected by the same problems encountered in the Wittig olefination of the diformylbiphenyl. However, to date, these have not been investigated. Diene 11 is converted in near quantitative yield to phenanthrene 10 in a ring closing metathesis reaction with the second generation Grubbs ruthenium carbene catalyst. Deprotection of the benzyl functionality and hydrogenation of the phenanthrene are simultaneously accomplished with 70 PSIG H2/Pd in 95% yield to give dihydrophenanthrene 20. Oxidation of 20 with diacetoxyiodobenzene (DAIB) affords masked ortho-benzoquinone 9 in 84% yield. Substitution occurs at the ortho position because of the resonance stabilization by the ortho methoxy of the oxidized cationic intermediate.
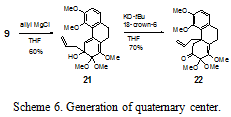
Allyl magnesium chloride addition to masked ortho-benzoquinone 9 affords tertiary alcohol 21 in 60% yield (Scheme 6). Tertiary alcohol 21 is deprotonated with potassium tert-butoxide in the presence of 18-crown-6 to induce an anionic oxy¬-Cope rearrangement to give 22 in a preliminary yield of 70%, installing the quaternary carbon center. The oxy-Cope rearrangement does not occur after the allyl Grignard reagent has been added because the potassium ion appears to be Lewis acidic enough to stabilize a degree of the negative charge on the alkoxide. In the Cope rearrangement, the 18 crown 6 generates a naked alkoxide ion which destabilizes the adjacent carbon–carbon bond, promoting the rearrangement.
Future work includes the ozonolysis of the terminal alkene of 22 to provide the precursor aldehyde to the reductive amination reaction. We postulate that the reduced steric hindrance at the carbon adjacent to the quaternary center, as compared to the model compound, should facilitate the ozonolysis of the terminal olefin. If, however, ozonolysis is ineffective, dihydroxylation/oxidative cleavage should provide the aldehyde in good yield. Reductive amination followed by Lewis acid mediated cyclization will afford the racemic natural product 3.
Discussion
The synthesis previously delineated yields the racemic mixture of hasubanonine. However, the stereochemistry of the molecule is controlled entirely at the point of allyl addition to the ortho-benzoquinone. This is so because the anionic oxy¬-Cope rearrangement is known to proceed on only one face of the molecule. In addition, the cyclization of the secondary amine onto the α,β-unsaturated ketal requires complete diastereoselectivity due to steric constraints. Therefore, an asymmetric synthesis of 3 can be accomplished by stereoselective allylation of benzoquinone 9. This will conceivably be achievable using a recently reported technique by Soderquist et al. involving a chiral allylborane reagent.7 If unsuccessful, other methods are known for the asymmetric allylation of ketones8 including TADDOL-modified Grignard reagents.9
Conclusion
Over the past year, I have synthesized a late stage intermediate in the total synthesis of hasubanonine. The current synthetic scheme employs standard organic chemistry functional group manipulation techniques as well as the methodology developed by our research group for the synthesis of acutumine. When the racemic synthesis of hasubanonine has been completed, the asymmetric allylation of the benzoquinone will be investigated as a means of controlling the stereochemistry of the two quaternary centers in the molecule.
References
- Douglas, C. J.; Overman, L. E. Proc. Natl Acad. Sci. U.S.A. 2004, 101, 5363.
- Reeder, M. D.; Srikanth, G. S. C.; Jones, S. B.; Castle, S. L. Org. Lett. 2005, 7, 1089.
- Tomita, M.; Ibuka, T.; Inubushi, Y.; Watanabe, Y.; Matsui, M. Tetrahedron Lett. 1964, 2937.
- Schultz, A. G.; Wang, A. J. Am. Chem. Soc. 1998, 120, 8259.
- Boger, D. L.; Borzilleri, R. M.; Nukui, S.; Beresis, R. T. J. Org. Chem. 1997, 62, 4721.
- Iuliano, A.; Piccioli, P.; Fabbri, D. Org. Lett. 2004, 6, 3711.
- Canales, E.; Prasad, G. K.; Soderquist, J. A. J. Am. Chem. Soc. 2005, 127, 11572.
- Denmark, S. E.; Fu, J. Chem. Rev. 2003, 103, 2763.
- Weber, B.; Seebach, D. Tetrahedron 1994, 50, 6117.