Darrell E. Hurt and Dr. Randall B. Shirts, Chemistry and Biochemistry
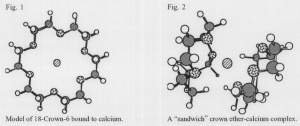
Since their discovery in 1967, crown ethers have proved a valuable tool for chemical separations of charged species that are otherwise inseparable.1 Crown ethers are a type of ligand, or a molecule that works by binding to another chemical species without breaking or forming chemical bonds. Crown ethers bind selectively – certain crown ethers bind strongly to certain species and not others. The selectivity of a crown ether is believed to be a function of both the size of its cavity and of the dihedral angle of the binding oxygen atoms.2,3 One can think of a crown ether as a molecular doughnut, (see figure 1) seeking its lost hole.
The classic example of a crown ether is 18-Crown-6. This ligand is a ring of 12 carbon atoms and six oxygen atoms symmetrically distributed about the ring, with the six electronegative oxygen atoms acting as the binding sites for an electropositive cation. (see figure 1) Several modeling techniques have been used to study 18-Crown-6.4,6 But no theoretical studies have been made for other crown ethers or for “sandwich” complexes of two ligands and one cation. (see figure 2) 1 used molecular mechanics to model these large systems of atoms.
Molecular mechanics (MM) is a classical method for modeling large chemical species. The method is analogous to building a model molecule from balls and sticks. The interactions between the balls and sticks are modeled using a force field. A force field is a collection of parameters describing the specific atoms in a molecule and of classical Newtonian equations that calculate the potential energy of the system depending on the positions of the atoms. By moving the atoms in the molecule and calculating the resulting potential energy, the computer can search for the lowest energy structure of the molecule. The geometry of the molecule in its lowest energy configuration can be thought of as a snapshot of the most common conformation of the molecule in the gas phase.
Before being able to calculate dissociation energies by minimizing molecular structures, I first had to add some parameters to the AMBER force field 5 for the alkaline earth metal cations. After parameterizing the force field for calcium, I did some preliminary calculations using I 9-Crown-6, 12- Crown-4, and several variations and “sandwich” complexes to determine the calcium cation dissociation energy. I would generate four hundred randomized models and then minimize their structures to find the absolute lowest energy conformation. Then I compared the energy of that minimum to that of the absolute lowest energy conformation of the free ligand. The difference in energy is the dissociation energy.
Our search scheme was designed to switch minimization algorithms at a certain threshold to maximize computer efficiency. However, it seemed that by switching algorithms halfway through the minimization, our results became nonsense. So we tried a simpler approach. When we got more nonsense we looked at other alternatives. To our chagrin, we found our force field had not been properly implemented in our software package! After correcting the implementation, I reparameterized the force field for calcium cations through trial-and-error and this time got some reasonable results. I presented these results during the Spring Research Conference of the College of Physical and Mathematical Sciences.
My results showed that the average energy penalty for detaching a binding oxygen atom from the cation is about 25 kcal/mol. The penalty is additive for the 1:1 complexes, so the dissociation energy of 18-Crown-6 is about 150 k caymol. However, the “sandwich” complexes exhibit some repulsion from the high concentration of electronegative oxygen atoms bound to the cation and so the energy penalty is not as great per detached oxygen.
While these results are reasonable, their drawback is that they use calculated partial charges while the AMBER force field is parameterized for an electrostatic potential fit derived from a high-level quantum mechanics calculation. Because the models are so sensitive to correct partial charges,6 my method of charge determination and guesswork parameters present some doubt to the accuracy of my results.
The greatest surprise in my results was the large number of local minima, or conformations, I was able to identify by minimizing randomized structures. We expected only one highly symmetric conformation to exist for each complex because of the strong electrostatic binding. Table I shows five low-energy minima for 12-Crown-4 bound to calcium. The 12-Crown-4 has relatively few minima because it is a smaller molecule and “sits” on top of the cation while the other complexes surround the cation, allowing more flexibility. I identified 32 minima in the 18-Crown-6 complex where all six oxygen atoms were still attached to the cation. The “sandwich” complexes had dozens of minima within an acceptable energy range. My work is continuing to determine how many unique conformations each complex can theoretically assume.
My most recent work has been to reparameterize the AMBER force field using a massive systematic search with increasingly fine search grids. I have determined the optimum parameters for each cation using partial charges derived from an ab initio quantum mechanics study on 18-Crown-6.4 I have also parameterized the POE force field7 for use with my models. Molecular mechanics assumes that parameters can be used on a class of molecules without having to change them for each system, so I can use these more accurate parameters for all of my model systems. I will continue my research this fall to find better dissociation energies using both force fields and using different distributions of partial charges.
Although I have been working on dissociation energies of crown ethers, my original grant from ORCA was to predict exchange rates for different crown ether complexes to compliment the work of David V. Dearden of the Department of Chemistry and Biochemistry.9 Exchange rates depend not only on the dissociation energy but also an energy of activation. In an attempt to model exchange rates, my work this summer has been concentrated on dynamics calculations at the University of Minnesota Supercomputer Institute. My work in Minnesota has cast some doubt on that goal, because exchange reactions do not include breaking or forming bonds. Since no bonds are broken or formed, the activation energy barrier that separates reactants and products is not very high. Without a large energy barrier, the intermediate structure and the reaction path between reactants and products is not well defined. The intermediate species in this case is just a high-energy conformation of the complex. Due to the large number of local minima I have identified with molecular mechanics alone, the reaction path will be difficult, if not impossible, to find, and the resulting exchange rates would be meaningless.9
1. C. J. Pederson, J Am. Chem. Soc. 1967, 89, 2495.
2. F. De Jong and D. N. Reinhoudt, Adv. Phys. Org. Chem. 1980, 17, 279.
3. E. D. Glendening et al., J Am. Chem. Soc. 1994, 116, 10657.
4. E. D. Glendening and D. Feller, J Am. Chem. Soc. 1996, 118, 6052, (ref 12-32.)
5. W. D. Cornell et al., J Am. Chem. Soc. 1996, 118(9), 2309.
6. R. B. Shirts and L. D. Stolworthy, J Inclusion Phen. 1994, 20(4), 297, (ref 11-23.)
7. G. D. Smith et al., J Phys. Chem. 1993, 97, 12752.
8. D. V. Dearden, unpublished results.
9. This research is supported by grants from the Department of Energy and National Science
Foundation. The aid of Joshua Harr and Randall B. Shirts is gratefully acknowledged
Table 1. Low energy minima of the 12-Crown-4 complex. The twelve torsion angles characterize the conformation. A – or + indicates a guache dihedral angle, while the 0 represents an anti- dihedral angle. The second conformation is almost 2 kcat/mol higher in energy and differs from the lowest energy conformation in the placement of the oxygen atom. Only the heavy atom dihedral angles are shown.