
James Coombs, Daniel Ess, Department of Chemistry and Biochemistry
Introduction
Heterodinuclear compounds containing a metal–metal bond represent a potentially useful subclass of catalyst in organic synthesis. Heterodinuclear compounds offer the possibility of increased reactivity due to interactions between metal centers (Scheme 1A). These so-called cooperative effects can enhance reactivity by changing the electronic density, increasing nucleophilicity/electrophilicity of the reactive metal, and by lowering the energy barriers for changes in oxidation state. These changes in reactivity can allow a heterodinuclear compound to be an effective catalyst for many chemical reactions that would be unfeasible with a mononuclear analogue.
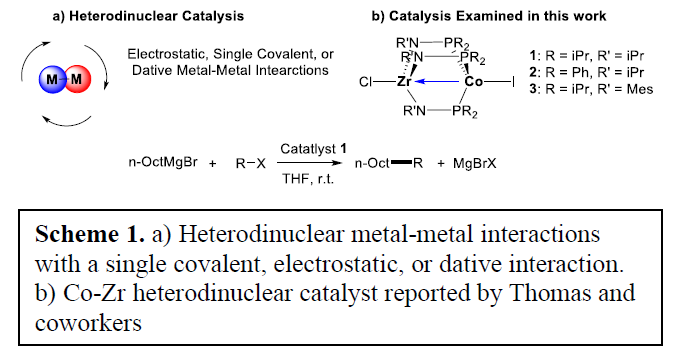 In recent years there has been resurgence in efforts to synthesize such compounds including several Co-Zr heterodinuclear compounds reported by Thomas and coworkers that catalytically performs Kumada coupling reactions (Scheme 1B). Thomas reported that the use of these heterodinuclear compounds resulted in moderate to high yields of C–C Kumada coupling products. This was in stark contrast to the Co-only analogue which produced only trace yields of product.
In recent years there has been resurgence in efforts to synthesize such compounds including several Co-Zr heterodinuclear compounds reported by Thomas and coworkers that catalytically performs Kumada coupling reactions (Scheme 1B). Thomas reported that the use of these heterodinuclear compounds resulted in moderate to high yields of C–C Kumada coupling products. This was in stark contrast to the Co-only analogue which produced only trace yields of product.
The development of Co based catalysts would allow for a larger application of Kumada coupling reactions to many synthetic processes. This prompted us to use density functional theory (DFT) to explain the efficiency of these compounds as Kumada coupling catalysts.
Methodology
All ground state and transition state structures were optimized in Gaussian 09 using the restricted and unrestricted M06L functional. The 6-31(d,p) basis set was used for H, C, N, O, Mg, and Cl. The LANL2DZ basis set and pseudopotentials were used for Co, Zr, Br, and I. Geometries were confirmed as minima or transition-state structures using normal-mode vibrational frequency analysis. Intrinsic reaction coordinate (IRC) calculations were performed to verify connections between transition states and intermediates. Conformational analysis and the evaluation of multiple spin states were completed for all structures. Electronic energies were calculated with the def2-TZVP//(U)M06-L//6- 31G(d,p)[LANL2DZ for Co, Br, Zr, and I] with an implicit SMD THF model and an ultrafine integration grid. Scheme 1. a) Heterodinuclear metal-metal interactions with a single covalent, electrostatic, or dative interaction. b) Co-Zr heterodinuclear catalyst reported by Thomas and coworkers
Results
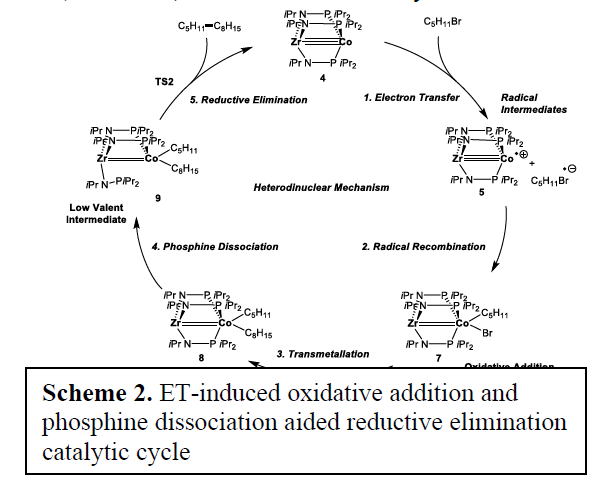
We decided to examine pre-catalyst 1 as it produced the highest yields. Our proposed catalytic cycle (Scheme 2) demonstrates the key role that the Co-Zr interaction plays in this catalysis. First, the presence of the Zr allows for two electron reduction of the pre-catalyst forming a potent electron donor species, 4, for electron transfer (ET). The resulting radical intermediates from the ET step can then recombine to give a formal oxidative addition. This then undergoes alkyl/bromide exchange between Mg and Co in the transmetalation step. Finally, the resulting dialkyl intermediate undergoes phosphine dissociation, thus controlling the electron density at Co, and allowing for energy efficient reductive elimination.
Discussion
By comparing to a Co-only analogue, we found that the Zr metal center assisted in this cycle in 3 crucial way: 1) The two metal centers allow for the formation of a doubly reduced species, 2) The electron withdrawing capacity of Zr allows for a lower reductive elimination barrier, 3) Zr provides scaffolding, holding the phosphine in place while it partially dissociates to form the low valent intermediate 9.
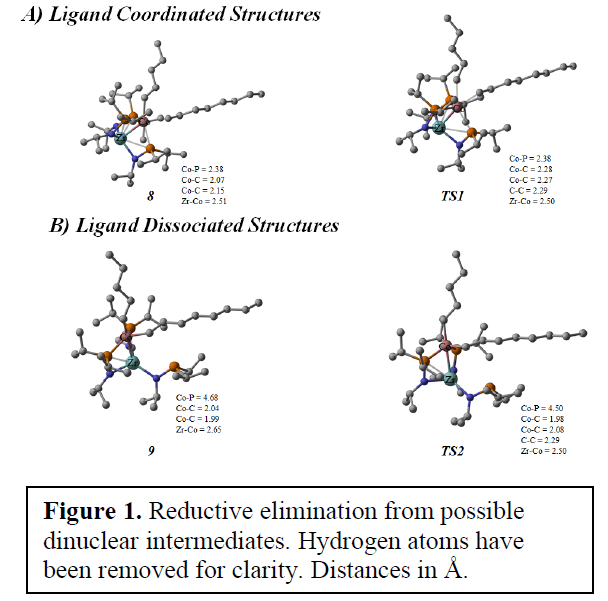
Figure 1 shows the structures we calculated that demonstrate the importance of the scaffolding provided by Zr. The energy calculated for TS1 was much too high to explain the fast room temperature reaction reported by Thomas. However, intramolecular phosphine dissociation allowed for optimal electron density of the Co center. This greatly reduced the calculated barrier for reductive elimination and has prompted Thomas to synthesize catalysts with fewer coordinated phosphines.
Conclusion
The metal-metal interaction in the heterodinuclear Zr-Co catalyst provides novel reactivity. The Zr-Co interaction allows for two electron reduction of the dinuclear pre-catalyst forming an electron rich active catalyst. This doubly reduced species allows for a low ET barrier facilitating the formal oxidative addition. In addition, the electron density is further optimized due to the intramolecular dissociation of phosphine. These significant differences allow for this heterodinuclear compound to be more effective than the Co only analogue in performing a Kumada coupling reaction.