Mark Woodbury and Dr. Scott Steffensen, Neuroscience Department
Introduction
Methamphetamine (METH) is a very powerful psychoactive drug that is highly addictive and toxic to the brain. Its use and abuse has been rising in recent years, and certain reports indicate that amphetamine-like drugs are the second most abused drugs worldwide. The side effects of METH abuse are varied and serious in nature, including: hyperthermia, tooth decay, heart attack, seizures, stroke, skin lesions, memory loss, confusion, anxiety, depression, psychosis, Parkinson’s Disease, and increased severity of HIV/AIDS (Chang et al., 2005; Curtin et al., 2015; Thanos et al., 2017). Despite these devastating effects, many of those addicted do not or cannot stop using and are finally met with fatal consequences.
METH is alleged to increase levels of dopamine in the brain in two ways: first, it decreases dopamine transporter (DAT) activity, resulting in decreased clearance of dopamine; second, METH increases synaptic dopamine release, possibly by recruiting additional dopamine vesicles during an action potential. This interesting second pathway may be more specific to METH than other psychostimulants and could be the reason for its devastating toxicity in the brain.
Research in the Steffensen lab has outlined a basic mechanism for the pathway by which METH increases dopamine in the brain, through direct activation of the sigma receptor. Indeed, pharmacological inactivation of the sigma receptor prevents METH-induced increases in synaptic dopamine release. The sigma receptor is involved in several different processes, and it was unknown how METH-induced activation of this receptor could influence dopamine release. However, it is known that the sigma receptor can activate the inositol triphosphate receptor (IP3R) which may influence intracellular processes. Activation of this receptor triggers intracellular calcium release which could in turn influence dopamine vesicle related machinery. Indeed, previous experiments which blocked intracellular calcium release decreased METH’s destabilizing effects on the cell. The aim of this study was to determine if IP3R activation was responsible for METH-induced changes in intracellular calcium.
Methods
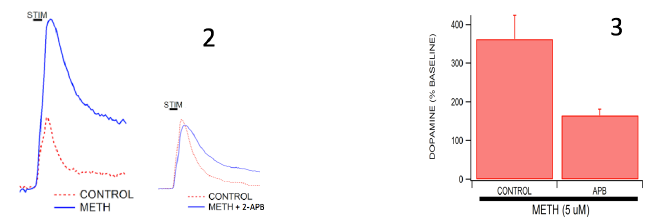
All experiments were performed on wild-type C57/Bl6 mice using a method called ex vivo voltammetry. Mice were anesthetized using isoflurane and decapitated according to approved IACUC protocols. Mouse brains were harvested and sectioned into 400 μm coronal slices. Slices containing the nucleus accumbens (NAc) were stored in oxygenated artificial cerebrospinal fluid (aCSF) for further use. These slices were then placed in bath perfusion chambers containing aCSF which were then placed in confocal microscopes. Fast-Scan Cyclic Voltammetry (FSCV) is used in our lab to measure dopamine transmission on a very rapid time scale (Ferris et al., 2013). This method allows us to stimulate and measure the release of dopamine in real-time within the brain slice. A stimulus electrode is placed adjacent to the carbon fiber recording electrode to stimulate an action potential and the release of dopamine, which is then recorded by the recording electrode. These electrodes are moved around in the NAc until a dopamine signal is found. Control collections are made by stimulating the electrode every 2 minutes until the dopamine release has stabilized to a fairly consistent amount upon stimulation. Once stabilized, the IP3R antagonist, 2-Aminoethoxydiphenylborate (2-APB), is bath applied at a concentration of 50 uM to the brain slice while still taking collections every 2 minutes. 2-APB has been shown to inhibit the extent of IP3R-induced calcium efflux from cerebellar microsomes (Bilmen and Michelangeli, 2002). Upon bath-application, signals are allowed to stabilize after the addition of 2-APB. Once the signal has stabilized again, METH is bath-applied by the same method at a concentration of 5 uM. The stimulated dopamine release is again recorded every 2 minutes until the signal is consistent and stable. This paradigm of experiment is compared to control experiments in which 2-APB is not applied to the brain slice and only METH applied and dopamine release is recorded in a similar manner.
Results
Our results indicate that neurons that are perfused with the IP3R antagonist, 2-APB, release significantly less dopamine upon METH administration compared to neurons that are not treated with 2-APB. METH alone produced an increase in dopamine release of 365% above baseline controls, whereas METH + 2-APB produced only a 161% increase from baseline controls.
Discussion/Conclusion
These results strongly implicate the involvement of the IP3R in the cascade of effects caused by METH which lead to the dysregulation of the neuron and an increase of dopamine efflux from the cell. The utilization of voltammetry to measure both evoked and basal release of dopamine gives strong support to our theory because it is perhaps the most effective method at comparing dopamine concentrations before and after drug administration. 2-APB is a very powerful agent to block the IP3R and also adds to the evidence towards this proposed mechanism.