
Marchant, Gilbert
Role of Sigma Receptors in Low-dose Methamphetamine Modulation of Dopamine Release in the Nucleus Accumbens
Faculty Mentor: Scott Steffensen, Psychology
Introduction
Methamphetamine (METH) is an addictive drug of abuse and METH use is a growing problem in the United States. The current dogma states that psychostimulants such as METH attack the mesocorticolimbic dopamine (DA) system, leading to abnormally high DA release when drugs of abuse are taken (Volkow et al., 2004). A key target area of the mesocorticolimbic system is the nucleus accumbens (NAc). Specifically, DA release in the NAc has been associated with reward and motivation (Oleson et al. 2012).
Recently, our research team proposed a new mechanism by which METH potentiates DA release via activation of the sigma receptor (σR) on the endoplasmic reticulum of presynaptic DA neurons in the NAc. The σR initiates a secondary messenger pathway culminating with an increase of superoxide, a reactive oxygen species. Superoxide interferes with the function of the vesicular monoamine transporter protein resulting in DA leaking into the cytosol. The increased DA concentration flows out through the dopamine transporter, a transmembrane protein in the presynaptic cleft, into the extracellular fluid where it binds both postsynaptic and presynaptic receptors leading to increased pleasure and reward.
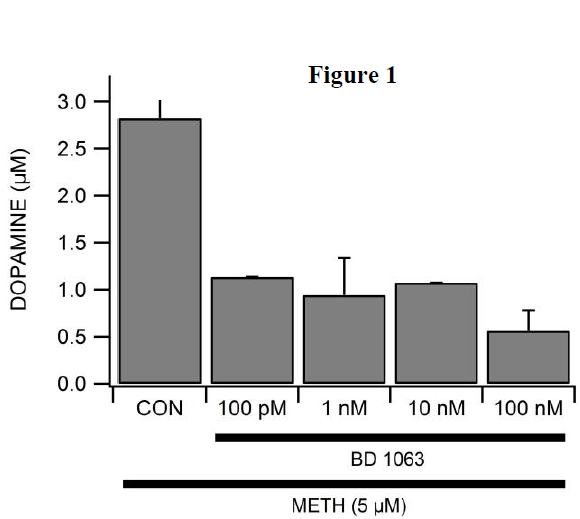
The role of the σR in this pathway required further elucidation. In this study, we studied the potency of BD1063, a σR antagonist, in blocking METH-induced DA release at several lower concentrations (100 nM, 10 nM, 1 nM, and 100 pM). We found that even these minute concentrations of BD1063 mitigated METH’s effects. In addition, we experimented with PRE-084 and DTG, σR agonists, but found that these molecules did not increase DA release.
Methodology
In this study, we utilized fast-scan cyclic voltammetry (FSCV), an electrophysiological method of measuring DA release in the brain. FSCV uses computer software in conjunction with carbon fiber microelectrodes to measure changes in electrical current resulting from the oxidation-reduction reaction of DA. Adult male mice were used in this study. Coronal brain slices were prepared, placed in an artificial cerebrospinal fluid (ACSF) bath, and a baseline DA signal was obtained. For more complete methods, see Schilaty et al., 2014.
We performed both phasic (electrically stimulated) and basal (non-stimulated) experiments with BD1063. In the phasic experiments, stimulations were performed every 2 min. BD1063 (100 nM, 10 nM, 1 nM, or 100 pM) was added to the ACSF bath followed by METH (5 μM). In the basal experiments, once the stimulated DA response was stable, the electrical stimulation was turned off and we performed a three hour recording. BD1063 was added after 30 minutes and METH was added after 60 minutes. For both PRE-084 and DTG, we performed phasic experiments to test a dose response (1 μM, 10 μM, 50 μM, 100 μM) followed by METH (5 μM). We used IBM SPSS Statistics 22 to analyze the results.
Results
We expected a decrease in the effectiveness of BD1063 in blocking METH-induced DA release as we decreased its concentration. However, BD1063 continued to mitigate DA release at 100 nM, 10 nM, 1 nM, and even 100 pM. These results are displayed in Figure 1.
Based on the theory that METH acts as a σR agonist, we hypothesized that adding σR agonists such as PRE-084 or DTG would augment DA release in the same manner as METH. However, we found that even large concentrations (100 μM) of these agonists failed to increase phasic DA release (PRE-084 (10 μM) decreased the baseline signal to 81.4% ± 4% of the baseline (p<0.001)).
Discussion
We were quite surprised at the potency of BD1063. The fact that a σR antagonist so effectively counteracts METH’s effects is further evidence that METH acts as a σR agonist in NAc neurons. The potential of incorporating a σR antagonist into a therapeutic agent for METH addicts is exciting. However, subsequent studies need to be performed in in vivo models to see if σR antagonists have similar physiological effects in behavioral experiments.
The fact that PRE-084 and DTG did not increase DA release like METH is concerning. If METH acts solely as a σR agonist, then these σR agonists should also potentiate DA release just as METH does. However, there are many confounding variables which could have caused this inconsistency. METH is known to enter the presynaptic neuronal terminals via the dopamine transporter (DAT) protein due to its similarity in structure to DA itself. Perhaps, these alternative σR agonists have no method by which to cross the cell membrane. In addition, it is theorized that METH actually reverses DAT when entering the neuron which causes DAT to pump DA out of the neuron in contrast to its normal role of DA reuptake. Thus, even if PRE-084 and DTG have a means by which to enter the neuron and agonize the σR, the unaltered DAT may be blocking the increased intracellular DA from exiting the neuron and entering the synaptic cleft. We need to experiment further with σR agonists in concert with DAT pharmacology to attempt to illuminate this problem.
Conclusion
The goal of this study was to understand more about the role of the σR in the mechanistic pathway by which METH leads to increased DA release in the NAc. Based on our results with BD1063, a σR antagonist, it seems clear that the σR is a definite target of METH and that METH acts to agonize this receptor. Admittedly however, our results with PRE-084 and DTG, σR agonists, proved useless in our attempts to understand this pathway. Further research should be performed to replicate our results of METH acting as a σR agonist as well as to further elucidate the downstream pathway which the σR then activates. We believe that understanding the actions of the σR will help to target therapeutic sites to aid in addiction recovery.