Steven Jones and Daniel Ess, Department of Chemistry and Biochemistry
Introduction
Cycloaddition reactions are key reactions used to construct complex cyclic scaffolds (large networks of carbon atoms that can be used in catalysis, synthesis, etc.). While classical reactions that form 6-membered rings are well-known and generally have a one-step reaction mechanism, 4-member ring-forming reactions are much less used, but could be very valuable for the construction of cyclobutane structures.
Tetrafluoroethylene (TFE) is used on a large scale by Dow and other chemical companies to form hydrofluoro compounds such as teflon. The current understanding of the mechanism of cycloaddition reactions with TFE is limited. Standard hydrocarbon reactions proceed via a welldocumented, concerted mechanism known as the Diels-Alder reaction; however, the addition of electron-withdrawing groups has been demonstrated to change the selectivity of these reactions, and the mechanism by which they proceed is as yet unknown. Specifically, cycloaddition between 1,3-butadiene and TFE results in exclusive formation of a 4 membered ring with a 2- carbon tail, as opposed to the 6 membered ring structure that is expected with similar cycloaddition reactions. Understanding the mechanism and periselectivity of these cycloaddition reactions will provide a long-standing resolution to many reactions and could lead to the development of new reactions with TFE.
Methodology
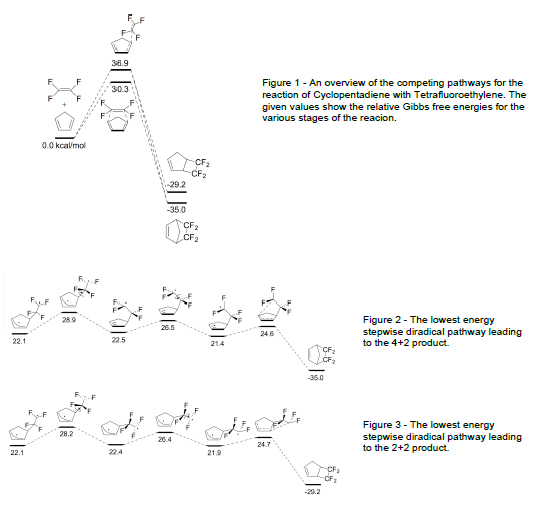
First, the one-step and multistep cycloaddition transition states for 1,3-butadiene and 1,3- cyclopentadiene cycloaddition with TFE were calculated. This was done using the program Gaussian 09 on the Fulton supercomputer. These calculations also served to help us determine a reasonable basis set to use for later dynamics calculations. All previous computational reports for the reaction of 1,3-butadiene and TFE were performed with simpler basis sets that contain no dynamical electron correlation effects. Our new results show that multistep and one-step pathways are competitive rather than the multistep diradical pathway being massively favored, as was previously proposed. We performed our direct dynamics study with the reaction of two TFE molecules to show whether the intermediate products formed thereby have a long or short lifetime. We then ran direct dynamics on the multistep radical pathways for 1,3-cyclopentadiene and TFE. Direct dynamics were run with the Singleton program ProgDyn hooked into Gaussian 09. We also used our own in-house C++ program for direct dynamics that is also hooked into Gaussian 09.
Results
We calculated and compared the energy barriers for hundreds of possible reaction mechanisms and conformations to determine the lowest possible pathway by which the reaction can proceed. Our calculations indicate that the stepwise diradical pathway for 2+2 addition is not thermodynamically favored, as has previously been suggested. Further, although the 4+2 product is not the primary product of the reaction, it is thermodynamically favored, indicating that the mechanism is the determining factor in the selectivity of the reaction. A summary of the energies and conformations calculated can be found in Figures 1–3. Unexpectedly, our dynamics calculations indicated very little influence on the reaction.
Conclusion
Our project has been very successful in examining the reaction we set out to characterize. We have performed a thorough investigation of the possible pathways and justifications for previously determined experimental data, and are confident in our assessment of the reaction energies and barriers. The reaction of TFE with CPD is a unique example of a vast array of cycloaddition reactions involving fluorocarbons, as well as other substituted electrophiles, and will undoubtedly lead to applications in diverse industrial syntheses.