Stokes, Ryjul
Quantifying the Efficiency of Heterobimetallic Complexes
Faculty Mentor: David Michaelis, Chemistry and Biochemistry
Transition metal catalysts have revolutionized the efficiency by which pharmaceuticals and other value added products are synthesized due to their unique ability to activate small molecules and drive the formation of new C-C and C-N bonds. In particular, the direct functionalization of olefins with transition metal catalysts is a powerful method of converting simple, cheap building blocks into more complex molecules. The challenge is that many ligands that control reaction selectivity often decrease the electrophilicity of the catalyst. Heterobimetallic complexes containing an early/late transition metal pair could provide a solution to this challenge by providing a new way to activate the late transition responsible for catalysis. The Lewis acidic early transition metal in these complexes can withdraw electron density from the late metal, making the latter more electrophilic, and thus more reactive towards olefin activation. It is the ability to add a second metal “ligand” to tune the electronic properties of the reactive center that makes heterobimetallic complexes a potentially superior alternative. Despite the obvious advantage of being able to tune the electronics of a catalyst independent of the metal system via this dative interaction, there are few examples of heterobimetallic mediated transitions.
Recently, Nagashima and coworkers reported a heterobimetallic catalyst that addressed many of the problems facing traditional monometallic catalysts. They synthesized a phosphinoamide ligand, what when combined with a TiCl4 or ZrCl4 yielded a stable early transition metal containing ligand. When heated with late transition metal salts, such as PtCl2 a heterobimetallic complex was formed. When assembled, there is a dative metal-metal interaction between the early and late metal centers. The Lewis acidic early transition metal is able to withdraw electron density from the second metal, causing it to become much more electrophilic that it would have been alone. Heterobimetallic complexes are advantageous due to their unique ability to tune the electrophilicity of the active metal center. By changing the different early and late transition metals, a range of electronic effects becomes possible. This allows for activation of the late transition metal in the presence of deactivating ligands. Nagashima was also able to show that the metal-metal interaction between the two metal centers would make ligands bound to the later transition metal more likely to nucleophilically attack electrophiles. In addition to the studies performed by Nagashima, Thomas and coworkers showed that altering the structure of the phosphinoamide ligand induced electronic effects, offering yet another way to electronically tune these complexes.
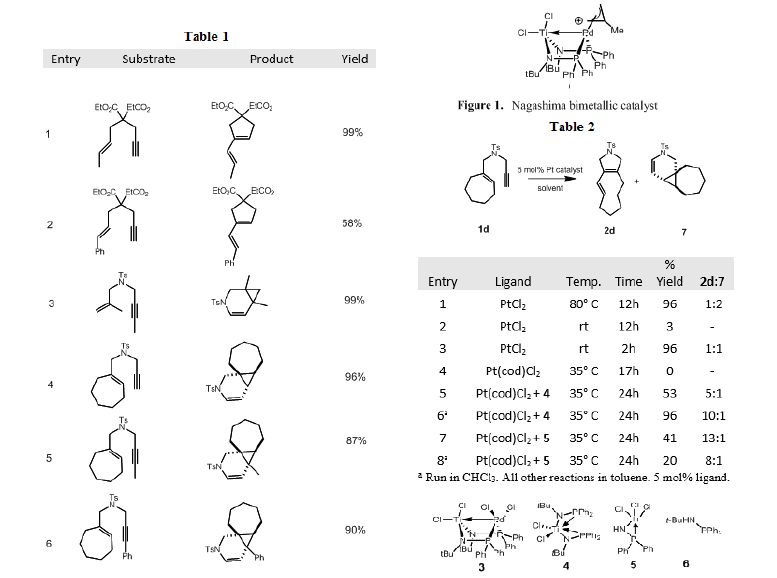
We decided to test the reactivity of Nagashima’s catalyst (Figure 1) as well as several novel catalysts that we developed in the cycloisomerization reaction of enynes. We noticed that in this particular reaction, our catalysts performed exceptionally well. Again, we believe that the exceptional reactivity observed is due to electron withdrawing ability of the metal ligand that speeds of the rate of nucleophilic addition. This reaction had been performed previously by Alois Fürstner, who reported that the reaction took up 12 hours at 80 °C, whereas our catalyst performed the reaction in only two hours at room temperature. Interestingly, the assembled heterobimetallic complex was inactive as a catalyst. The active catalytic species in this reaction is the complex formed with PtCl2 and ligand 5. Unfortunately, we were not able to isolate the platinum bound monodentate species, and 31P NMR studies proved inconclusive. To confirm our hypothesis that it was indeed the monodentate Pt-Ti complex, we added two equivalents of ligand 5 and observed a drop of 10% in the overall yield of the reaction. This confirmed that the monodentate species is the active catalyst. We believe that upon higher catalyst loading, bidentate ligand forms in situ which slows the reaction. Several other reactions with various common ligands in the presence of PtCl2 showed poor yield or no reaction, confirming the necessity of the Lewis acidic titanium ligand.
With optimized conditions in hand, we tested several diethylmalonate and tosyl protected enyne substrates (Table 1). During the course of the study, we notice that though we were attaining excellent yields in the majority of the reactions, the observed selectivity was different than that reported by Fürstner. In fact, the selectivity that we observed seemed to favor the more strained product (Table 2). Thus, the ability to perform this reaction in situ and modify the ligands used allows us the opportunity to drive reaction selectivity to the desired product.
In conclusion, heterobimetallic complexes served to be excellent catalysts for the electrophilic activation of alkynes in the cycloisomerization reaction of enynes, and we look forward to testing other reactive systems with these catalysts. We hope that this work can improve efficiency in key pharmaceutical bond forming reactions in order to decrease production costs and increase accessibility to important medications.