
Lyman, Cole
Utilizing the Power of Graphical Processing for DNA Mapping: a Comparison of GNUMAP and BarraCUDA
Faculty Mentor: Mark Clement, Department of Computer Science
Introduction
Recent advances in genome sequencing technologies have resulted in a large increase
in the amount of genetic data available. Large Genome Wide Association Studies
(GWAS) have the potential to identify the causes of cancer, Alzheimer’s disease, heart
failure and many other diseases if the large quantities of data that are becoming
available can be analyzed effectively. Next-generation read mapping software, a crucial
step in analyzing genetic data, is slow while trying to achieve high mapping accuracy.
One approach to speeding up next-generation read mapping focuses on using
Graphical Processing Units (GPUs). This project compared the effectiveness of two
genome mappers, GNUMAP1 and BarraCUDA2, to measure the impact of GPUs on
genome mapping.
Methodology
To compare the effectiveness of these two genome mappers we measured how long
they took and how accurate they were when mapping a real dataset and a synthetically
generated dataset. The real dataset of human reads was obtained from the 1000
Genomes3 dataset as well as a set of synthetic reads generated by ART4 . Both sets of
reads were aligned to the hg19 human genome assembly. The total time for alignment
was measured and the alignments were compared to measure accuracy.
The Fulton Supercomputing Lab’s Mary Lou supercomputer was used to run both
programs. Nvidia’s GK210 GPUs were used to run Barracuda, and Intel’s 12-core
Haswell (2.3 GHz) processors were used to run both GNUMAP and Barracuda.
Results
When mapping the 1000 genomes dataset, GNUMAP and BarraCUDA took a
comparable amount of time with BarraCUDA being faster than GNUMAP. Both aligners
were able to align about 90% of the 1000 genomes reads, with BarraCUDA aligning
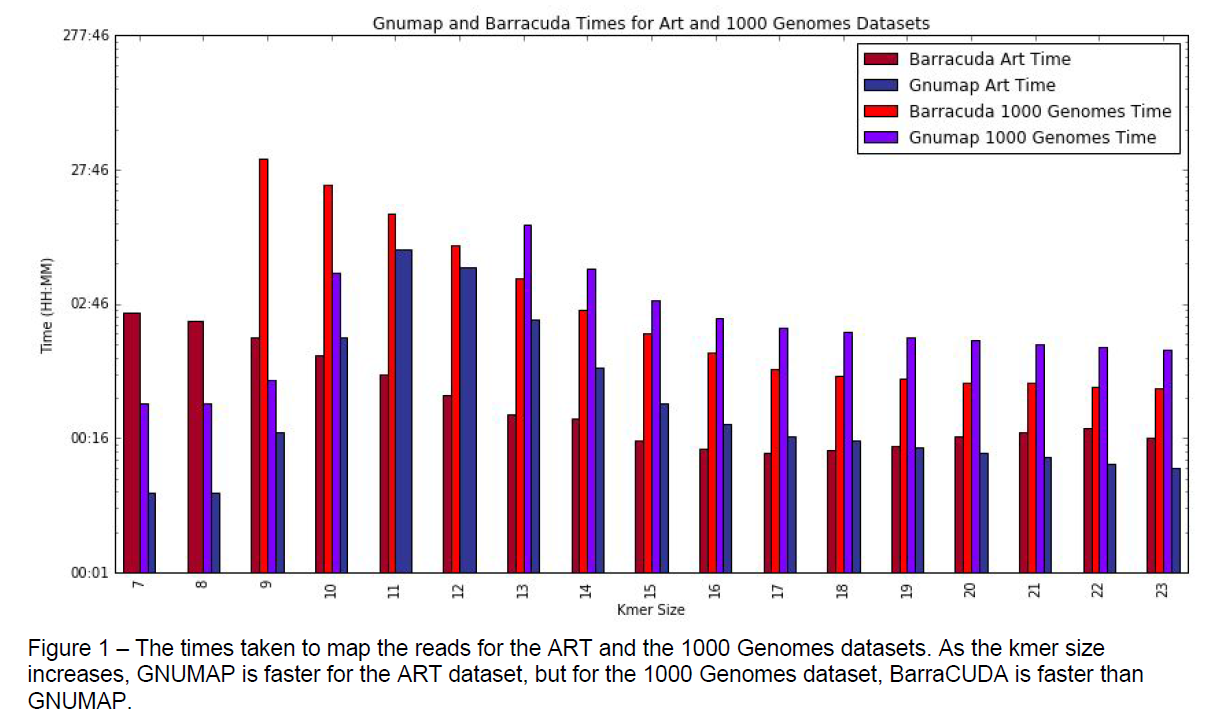
about 1% more than GNUMAP. When aligning the synthetic reads BarraCUDA was also
faster for smaller kmers, but GNUMAP’s time falls as the kmer size increases.
GNUMAP and BarraCUDA were able to align a comparable amount of the synthetic
reads, with BarraCUDA aligning a higher percentage of reads to the correct genomic
position.
Discussion
With the times and accuracies of GNUMAP and BarraCUDA being comparable, even
though BarraCUDA uses a GPU which has thousands more threads than a CPU, which
GNUMAP uses. One explanation for why BarraCUDA is not as fast as expected is the
high cost of data transfer to and from the GPU. Data transfer to and from the GPU takes
a relatively long time when transferring larger datasets, such as read and genome data.
While there may be a time advantage of using the GPU in read alignment, the cost of
transferring the data eliminates any significant reduction in time.
Conclusion
We conclude that BarraCUDA is a viable read mapper for those that have access to a
high-end GPU, which can be a more cost efficient solution than a high performance
computing cluster, that programs like GNUMAP usually run on. With the marginal
speedups gained by BarraCUDA there is no loss in accuracy and is in some cases
more accurate than GNUMAP. If improvements on the data transfer efficiency for GPUs
are made, then read mapping could become significantly faster on GPUs than on CPUs.