
Dayton, Jonathan
Using bioinformatics to increase the number of tumors that can be treated with existing drug treatments
Faculty Mentor: Stephen Piccolo, Biology
Introduction
Typically, tens or even hundreds of mutations are observed in the DNA of a single
tumor by the time it has been detected1-4. Knowledge of these mutations may be useful
in guiding the way the tumor is treated. In some cases, if a tumor has a mutation in a
certain gene, this may indicate that the tumor can be treated with a certain drug. For
example, the drug Trastuzumab is a targeted therapy for breast cancer patients with
mutations in the HER2 gene5. In other cases, a mutation may be able to be targeted by
a drug even if the drug wasn’t developed specifically for that mutation6. For both cases,
many such relationships have been identified linking mutations to treatments7.
However, many tumors do not contain a mutation with a known treatment. Because
there are numerous genes that may be mutated in a tumor, it may be economically
infeasible to develop targeted therapies for every rare mutation. However, we may be
able to reuse existing drugs to target rare mutations by identifying similarities between
tumors that harbor rare and common mutations.
Our research focused on identifying similarities between tumors by measuring the
degree of similarity between the downstream effects of different mutations. We
hypothesized that if the RNA expression data was very similar for two tumors with two
different mutations, it would indicate that the two mutations could potentially be targeted
by the same treatments.
Methodology
We used publicly available data from The Cancer Genome Atlas (TCGA)8. We acquired
data representing DNA mutations and RNA transcription for 9,300 tumors across 25
cancer types. We filtered the data using the Python programming language9 in order to
exclude mutations that were unlikely to play a role in tumor development. We also
batch adjusted10 the RNA data based on cancer type in order to make comparisons
between the 25 different cancer types.
We assigned tumors to groups based on which mutations the tumors had. We used a
Random Forests machine learning algorithm11 to analyze similarities between tumors in
the different groups. We first performed this analysis on tumors from specific cancer
types and then expanded the analysis to include data from 25 cancer types. We wrote
our analytical pipeline using the R programming language12.
We compared our results against known gene-drug relationships using the data found in
the Clinical Interpretations of Variants in Cancer (CIViC) database7 in order to assess
the consistency of our results with prior knowledge.
Results
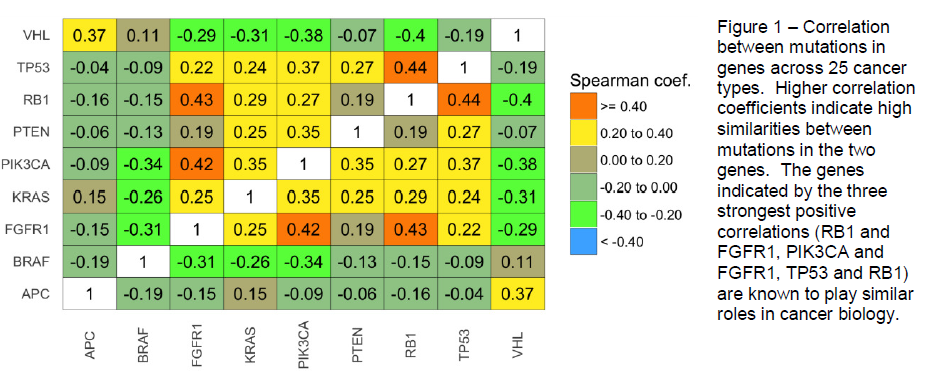
We performed our analysis across 25 cancer types on the 9 genes that were mutated in
a mutually exclusive manner in at least 50 tumors. Several interesting relationships
between genes were apparent, including a strong correlation between the TP53 and
RB1 genes, which play critical roles in regulation of the cell cycle and DNA repair13,14.
FGFR1 was strongly correlated with PIK3CA, likely due to FGFR1’s role as an upstream
activator of PIK3CA15. FGFR1 was also strongly correlated with RB1, and it has been
shown recently that these two genes are regulated by the same microRNA16.
We then used the same classification algorithm on genes that were mutated less
frequently. Using a minimum threshold number of five mutated samples, we found two
relatively strong correlations. The first was between VHL and MTOR1, which play
interconnected roles in renal cell carcinoma17. The second correlation was between
NRAS and BRAF. These genes interact directly with each other via the Ras → Raf →
MEK → ERK cascade15. Also, the CIViC database indicates that several antibodybased
treatments—including Cetuximab, Selumetinib, and Vemurafenib—target tumors
with mutations in either of these genes.
Conclusion
In many cases, the transcriptional patterns were highly predictive of mutation status,
thus indicating that, in many cases, individual mutations influence transcription in
distinct ways. Using lower-frequency genes that had been excluded from the initial
analysis, we identified genes that, when mutated, resulted in transcriptional patterns
similar to some of the genes from our initial set. In several cases, these similarities
coincided with prior knowledge about cancer biology as well as with known gene-drug
relationships. Our findings validate our analysis as a way to identify pairs of genes that,
when mutated, may serve as biomarkers for the same treatment. These observations
show promise to help pharmacologists and clinical trialists narrow the search space for
candidate gene-drug therapeutic associations.
References
1 S. Jones, X. Zhang, D. W. Parsons, J. C.-H. Lin, R. J. Leary, P. Angenendt, P. Mankoo, H. Carter, H. Kamiyama, A.
Jimeno, S.-M. Hong, B. Fu, M.-T. Lin, E. S. Calhoun, M. Kamiyama, K. Walter, T. Nikolskaya, Y. Nikolsky, J.
Hartigan, D. R. Smith, M. Hidalgo, S. D. Leach, A. P. Klein, E. M. Jaffee, M. Goggins, A. Maitra, C. Iacobuzio-
Donahue, J. R. Eshleman, S. E. Kern, R. H. Hruban, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V.
E. Velculescu, and K. W. Kinzler, “Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global
Genomic Analyses,” Science (80-. )., Sep. 2008.
2 W. W. Parsons, J. C.-H. C. Lin, S. Jones, I.-M. M. Siu, X. Zhang, A. A. Rasheed, S. K. N. K. Marie, R. J. Leary, S.
M. O. M. Shinjo, P. Angenendt, P. Mankoo, H. Carter, G. L. Gallia, A. Olivi, R. McLendon, S. Keir, T. Nikolskaya, Y.
Nikolsky, D. A. Busam, H. Tekleab, L. A. Diaz, J. Hartigan, D. R. Smith, R. L. Strausberg, H. Yan, G. J. Riggins, D. D.
Bigner, R. Karchin, N. Papadopoulos, G. Parmigiani, B. Vogelstein, V. E. Velculescu, and K. W. Kinzler, “An
integrated genomic analysis of human glioblastoma multiforme.,” Science, vol. 321, no. 5897, pp. 1807–1812, Sep.
2008.
3 T. C. G. A. R. Network, “Comprehensive molecular profiling of lung adenocarcinoma.,” Nature, vol. 511, no. 7511,
pp. 543–50, Jul. 2014
4 The Cancer Genome Atlas Research Network, “Comprehensive genomic characterization of squamous cell lung
cancers.,” Nature, vol. 489, no. 7417, pp. 519–25, Sep. 2012.
5 M. A. Molina, J. Codony-Servat, J. Albanell, F. Rojo, J. Arribas, and J. Baselga, “Trastuzumab (Herceptin), a
humanized anti-HER2 receptor monoclonal antibody, inhibits basal and activated HER2 ectodomain cleavage in
breast cancer cells,” Cancer Res., vol. 61, no. 12, pp. 4744–4749, 2001.
6 M. J. Garnett, E. J. Edelman, S. J. Heidorn, C. D. Greenman, A. Dastur, K. W. Lau, P. Greninger, I. R. Thompson,
X. Luo, J. Soares, Q. Liu, F. Iorio, D. Surdez, L. Chen, R. J. Milano, G. R. Bignell, A. T. Tam, H. Davies, J. a.
Stevenson, S. Barthorpe, S. R. Lutz, F. Kogera, K. Lawrence, A. McLaren-Douglas, X. Mitropoulos, T. Mironenko, H.
Thi, L. Richardson, W. Zhou, F. Jewitt, T. Zhang, P. O’Brien, J. L. Boisvert, S. Price, W. Hur, W. Yang, X. Deng, A.
Butler, H. G. Choi, J. W. Chang, J. Baselga, I. Stamenkovic, J. a. Engelman, S. V. Sharma, O. Delattre, J. Saez-
Rodriguez, N. S. Gray, J. Settleman, P. A. Futreal, D. a. Haber, M. R. Stratton, S. Ramaswamy, U. McDermott, and
C. H. Benes, “Systematic identification of genomic markers of drug sensitivity in cancer cells,” Nature, vol. 483, no.
7391, pp. 570–575, Mar. 2012.
7 M. Griffith, N. C. Spies, K. Krysiak, A. C. Coffman, J. F. McMichael, B. J. Ainscough, D. T. Rieke, A. M. Danos, L.
Kujan, C. A. Ramirez, A. H. Wagner, Z. L. Skidmore, C. J. Liu, M. R. Jones, R. L. Bilski, R. Lesurf, E. K. Barnell, N.
M. Shah, M. Bonakdar, L. Trani, M. Matlock, A. Ramu, K. M. Campbell, G. C. Spies, A. P. Graubert, K. Gangavarapu,
J. M. Eldred, D. E. Larson, J. R. Walker, B. M. Good, C. Wu, A. I. Su, R. Dienstmann, S. J. Jones, R. Bose, D. H.
Spencer, L. D. Wartman, R. K. Wilson, E. R. Mardis, and O. L. Griffith, “CIViC: A knowledgebase for expertcrowdsourcing
the clinical interpretation of variants in cancer.,” 2016.
8 Available from https://gdc.cancer.gov/.
9 Python Software Foundation, “Python Language Reference, version 2.7,” Python Software Foundation. 2013.
10 W. E. Johnson, C. Li, and A. Rabinovic, “Adjusting batch effects in microarray expression data using empirical
Bayes methods.,” Biostatistics, vol. 8, no. 1, pp. 118–27, Jan. 2007.
11 L. Breiman, “Random Forests,” Mach. Learn., vol. 45, no. 1, pp. 5–32, Oct. 2001.
12 R Core Team, “R: A Language and Environment for Statistical Computing.” Vienna, Austria, 2016.
13 M. L. Smith and Y. R. Seo, “p53 regulation of DNA excision repair pathways.,” Mutagenesis, vol. 17, no. 2, pp.
149–156, 2002.
14 R. Cook, G. Zoumpoulidou, M. T. Luczynski, S. Rieger, J. Moquet, V. J. Spanswick, J. A. Hartley, K. Rothkamm, P.
H. Huang, and S. Mittnacht, “Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous
end-joining,” Cell Rep., vol. 10, no. 12, pp. 2007–2019, 2015.
15 M. Tanabe and M. Kanehisa, “Using the KEGG database resource,” Curr. Protoc. Bioinforma., no. SUPPL.38,
2012.
16 P. Rodriguez-Otero, J. Román-Gómez, A. Vilas-Zornoza, E. S. José-Eneriz, V. Martín-Palanco, J. Rifón, A. Torres,
M. J. Calasanz, X. Agirre, and F. Prosper, “Deregulation of FGFR1 and CDK6 oncogenic pathways in acute
lymphoblastic leukaemia harbouring epigenetic modifications of the MIR9 family,” Br. J. Haematol., vol. 155, no. 1,
pp. 73–83, 2011.
17 B. Kucejova, S. Peña-Llopis, T. Yamasaki, S. Sivanand, T. A. T. Tran, S. Alexander, N. C. Wolff, Y. Lotan, X.-J.
Xie, W. Kabbani, P. Kapur, and J. Brugarolas, “Interplay Between pVHL and mTORC1 Pathways in Clear-Cell Renal
Cell Carcinoma.,” Mol. Cancer Res., vol. 9, no. 9, pp. 1255–1265, 2011.