
Anna Guyer and Stephen Piccolo, Department of Biology
Introduction
Biomedical data are increasing in size and complexity. To make sense of these data,
biomedical researchers often use “machine-learning” algorithms, which are developed by
the computer-science community. Our goal was to perform a systematic comparison of
many of these algorithms across 100 data sets to identify which algorithms perform best
for this type of data. To help meet this goal, we also planned to carefully curating data from
the public domain for others to use in their own comparisons.
Unlike DNA, which changes little from cell to cell, gene-expression levels vary dramatically
across different types of tissues and under different conditions. Because of this variance,
gene expression data can often be used to predict biomedical outcomes. Such outcomes
might include development of a disease, survivability, reaction to a drug, and other such
medically relevant information. There are a wide range of “machine learning” algorithms
that researchers use; because human health is at stake, having the utmost accuracy is
important. However, because of the overwhelming number of algorithms available,
researchers often use whatever algorithm(s) they have used previously, even though there
may be a more accurate alternative.1 We hypothesized that certain algorithms will perform
better than others overall and that some algorithmic attributes may be best suited for
certain dataset characteristics.
Methods
Previous studies have shown that many factors influence how well a classification
algorithm performs, and we set out to find which conditions work best with various
algorithms. We first compiled a uniform collection of datasets and curated them into a
consistent, usable format. We collected raw data from Gene Expression Omnibus (GEO),
a public repository supported by the National Center for Biotechnology. We searched for
datasets that represent a variety of biomedical outcomes and that were profiled using the
same gene-expression technology (for consistency). We downloaded each dataset and
extracted relevant clinical information and further filtered the list of datasets. This was done
by both hand curation by writing computer scripts to download and reformat the data. We
then normalized the expression values using the SCAN.UPC package2. This package
enables normalization on a single-sample level, allowing for no dependence upon other
samples. This package also makes it possible to map the data to genes2. For some
studies, we had to correct for batch effects, which occur when some samples are
processed at different times than other samples and thus may introduce biases. To do this,
we used ComBat, another R package3. Once we reached the desired uniformity, we
applied 24 classification algorithms to each dataset. Lastly, we analyzed and compared the
performance of each algorithm to determine which algorithms are most accurate.
Results and Conclusions
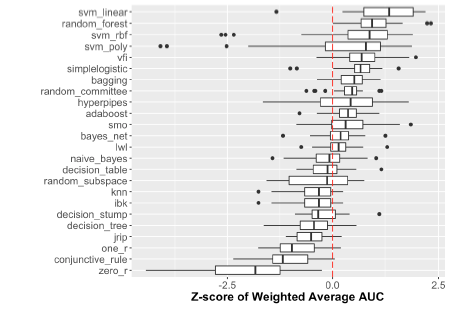
We performed an analysis including an initial 24 algorithms and 30 dataset/class combinations. Each dataset/class combination was run on each algorithm across 100 iterations. We found that performance varied depending upon the outcome being predicted. Some types of outcomes (such as tumor/normal status) were predicted more accurately than others (such as disease stage/survival). We also compared the
performance of each algorithm across all dataset/class combinations using z-score normalized accuracy value (see Figure 1). We found a trend that Support Vector Machines (SVM Linear, SVM rbf, SVM poly) and Random Forest (RF) algorithms outperformed most other algorithms in their accuracy of prediction, as would be expected by the scientific community, though there were iterations in which these algorithms performed very poorly. It was also interesting to find that Voting Feature Intervals (VFI) and Simple Logistic had similar performance to these widely accepted “best” classification prediction algorithms. VFI was more consistent in its performance with fewer low-performing outlier iterations. This suggests that VFI may be a reliable algorithm for gene expression related classification and it would be of benefit to the scientific community to put this algorithm into wider use.
This research is only a start. The tools and methods of downloading and filtering this data
from the public repository will enable others—and ourselves—in future studies. We quickly
discovered that there are often discrepancies in the formats of datasets. We also
recognize that in most projects there is a level of time sensitivity as to how quickly the
analysis needs to be done and in some cases the extra time needed for an algorithm to
run can more than negate the benefit of a little added accuracy. We would like to
investigate this in the future to evaluate accuracy vs. time and perform a cost/benefit
analysis. We only scratched the surface in the comparison of these algorithms, and work is
still in progress to expand the number of datasets and algorithms used. This research was
presented at the annual conference for the International Society for Biocuration, and a
manuscript will soon be submitted to an academic journal about this study.
[1] Fernández-Delgado M, Cernadas E, Barro S, et al. 2014. Do we Need Hundreds of
Classifiers to Solve Real World Classification Problems? Journal of Machine Learning
Research. 15(2014) 3133-3181.
[2] Piccolo, S. R. et al. A single-sample microarray normalization method to facilitate
personalized-medicine workflows. Genomics 100, 337–344 (2012).
[3] Johnson, WE, Rabinovic, A, and Li, C (2007). Adjusting batch effects in
microarray expression data using Empirical Bayes methods. Biostatistics 8(1):118-127.