
Nathan Clement and Dr. Quinn Snell, Computer Science
Introduction
In order to create and maintain a healthy human body, over three billion nucleotides (molecules of DNA, collectively called a genome) need to work together in harmony. Each cell in the human body needs its own copy of the genome, so during growth an individual’s DNA is replicated many times. Because the process of replication is not perfect, there are occasional mistakes. Sometimes, these mistakes can be fixed by the body’s own repair mechanisms, but occasionally, a single change in one of the three billion molecules in a human genome have lifethreatening results (for example, see the entry on Cystic Fibrosis at http://www.ncbi.nlm.nih.gov/omim/219700). In addition, certain changes (called Single Nucleotide Polymorphisms or SNPs, pronounced “snip”) that are passed on genetically do not have physical manifestations, but can cause a predisposition to a specific disease later in life. Much research over the past decade has been aimed toward first, identifying SNPs in different individuals, and then second, linking them with diseases.
Since the completion of the Human Genome Project in 2001, researchers have been able to more closely study an individual genome and changes between different genomes. Scientists have been able to use ever-advancing technology to determine the exact DNA sequence of an individual in a few weeks, with the hopes of reducing this to 15 minutes in the near future. Currently, the bottleneck for linking SNPs to a disease lies in finding which of the three billion nucleotides in a genome are changed—and which changes are important.
Sequence Mapping and SNP Calling
At the root of this bottleneck lies a process called sequence mapping. Employing a technology called Next-Generation Sequencing, the entire human genome is broken into fragments (ranging in size from 35 to 500 bases) that are then sequenced and passed to a computer. It is then the job of a computer program to determine to where, in a previously existing reference genome, each fragment matches best. When the mapping process is complete, the computer program will provide the researcher with a list of changes that do not appear in the reference genome.
As can be imagined, there are difficulties in this process. Next-Generation Sequence converts vast amounts of biological DNA into computer bits, but occasionally this process is flawed, introducing false mutations into the data. The biggest challenge is identifying the difference between actual SNPs that cause disease and spurious changes resulting from a faulty sequencing or mapping process.
Results
Over the past two years, we developed a program called GNUMAP, which more accurately maps short DNA fragments to a human genome. Since fall of 2009, our efforts have focused on increasing the functionality of GNUMAP to include a SNP calling method. In order to do this, we employed the use of complex statistical models, high-performance computing methods, and BYU’s own supercomputer. In the end, we showed that our method was much better than the leading programs.
The current best methods for making SNP predictions from Next-Generation Sequencing data are MAQ2 and SOAP-SNP3. In order to test GNUMAP against these programs, we modified an existing genome at pre-defined locations, simulated a sequencing process, and compared the results each program achieved with what was expected. Since speed is critical to this process, we also recorded the time each program took. In Table 1, true positives are defined as locations in the simulated genome that were correctly called as SNPs; false positives are those that were called SNPs that should not have been; and false negatives are locations that were not called as a SNP that should have been.
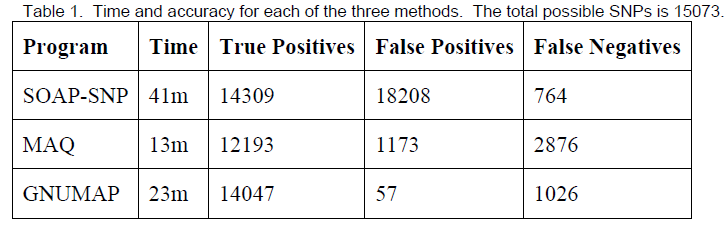
As can be seen in Table 1, GNUMAP is able to outperform both SOAP-SNP and MAQ, especially in the low number of false positives. While SOAP-SNP correctly identified more mutated locations (94.9%), the enormous number of false positives it reported makes it nearly impossible to identify a correct SNP.
Conclusion
Based on the preliminary results from the simulated data set, we can conclude that GNUMAP is more selective at finding SNPs without losing accuracy. In addition, the statistical model we use to differentiate between SNPs and noise provides the researcher with a p-value corresponding to the probability that a mutation has been correctly identified.
Currently, we are working to identify known and unknown SNPs in a real human dataset and compare the results with MAQ and SOAP-SNP. This ongoing research is part of a paper that will soon be published. In addition, our method is being used in several real-world applications at the Huntsman Cancer Institute, and has been downloaded in several locations around the world.
References
- Li H., Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality
scores. Genome Research 2008, 18:1851–1858 - Li R, Yingrui L, Fang X, Yang H, Wang J, Kristiansen K, Wang J: SNP detection for massively parallel
whole- genome resequencing. Genome Research 2009, 19: 1124–1132.