
Susie Choi and Dr. Young Wan Ham
Abstract
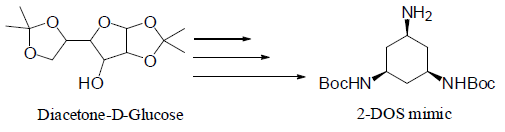
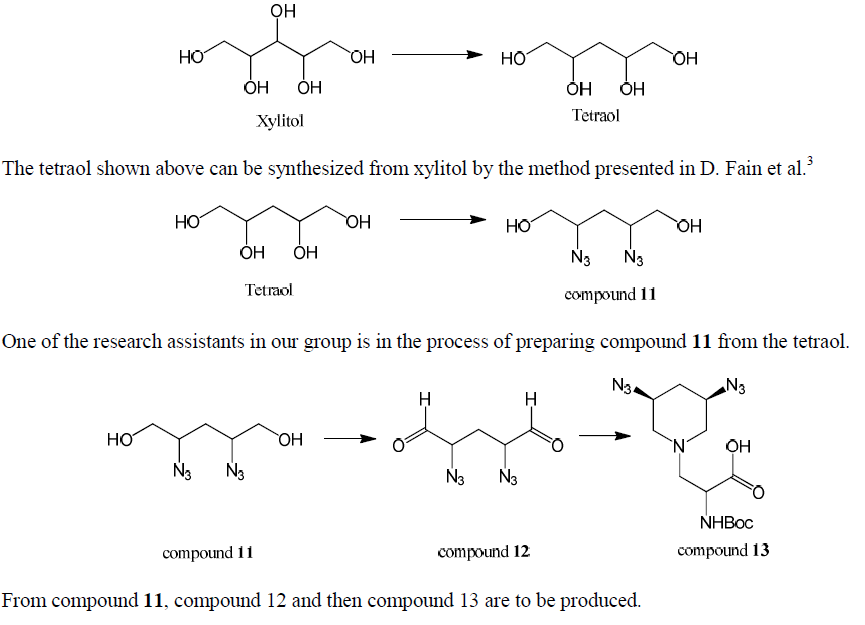
2-deoxystreptamine (2-DOS) mimic was to be synthesized in several steps from commercially available diacetone-D-glucose in an effort to prepare novel amino acid residues for antiviral therapeutic development. The method presented in this paper was given up due to unexpected problems occurring at three of the 10 steps. A new method, which uses xylitol as the starting material and takes fewer steps, is to be used.
Introduction
The virally encoded Rev protein of HIV-1 plays a critical role in viral replication by regulating the transport of unspliced and partially spliced viral RNA from the nucleus to the cytoplasm of infected cells.1 Rev protein binds to a specific region of the HIV-1 mRNA, called the Rev Responsive Element (RRE), to mediate this transport. The interaction between Rev protein and the RRE of HIV mRNA is essential for viral replication, yet binding of Rev protein to RRE lacks specificity. This is because their interaction relies heavily on electrostatic interactions between Rev protein’s basic amino acid residues and the negatively charged phosphate backbone of the RNAs. Our group expect that unnatural oligopeptides that bind to the RRE with a higher specificity than natural viral Rev protein block the RRE of the natural Rev protein, therefore, stalling viral replication in HIV-infected cells.
The goal of my project was to synthesize a 2-DOS mimic that was to be incorporated onto the side chains of certain amino acids to produce novel amino acid residues (Fmoc-NH-Z-OH). 2-DOS (Figure 1) has been shown to recognize 5’-GU-3’ and possibly 5’-GG-3’ base steps in a sequence-specific manner in bacterial RNAs through highly conserved hydrogen bonds2. Therefore, we propose that the 2- DOS mimic be prepared to recognize 5’-GU-3’ and 5’-GG-3’ dinucleotides in the binding site of HIV RRE. We expect that the oligonucliotides containing Fmoc-NH-Z-OH will improve binding specificity towards RRE in HIV mRNA.
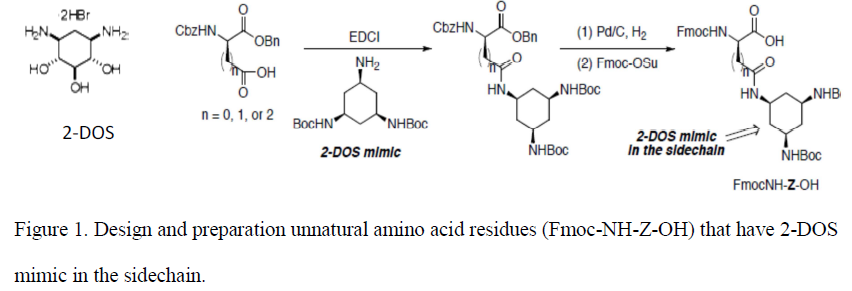
Experimental Section
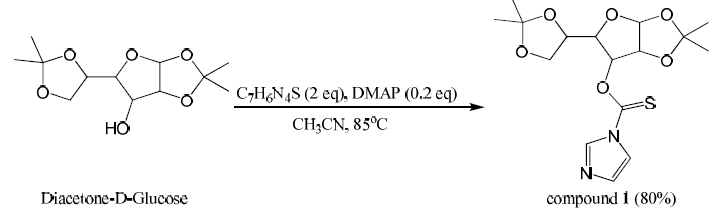
Diacetone-D-Glucose (1.0 equiv, 19.21 mmol) was dissolved in acetonitrile (40 mL) in a dry round bottom flask. DMAP (0.2equiv, 3.84 mmol) and 1,1′-Thiocarbonyldiimidazole (2 equiv, 38.42 mmol) were added. This was refluxed and stirred for 60 minutes at 85 ⁰C until the starting material had been consumed. Acetonitrile was evaporated off and the crude product was extracted three times with dichloromethane. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated. The product was purified by column chromatography (50% ethyl acetate/hexanes) producing compound 1 (80% yield). Mass spectroscopy confirmed the presence of a major peak at 370 m/z (compound 1).
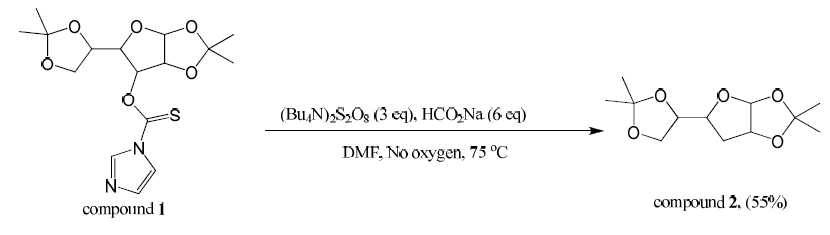
Compound 1 (1.0 equi, 15.37 mmol) was dissolved in dimethylformamide (70 mL) in a dry round bottom flask. Sodium formate (6 equiv, 92.21 mmol ) and (Bu4N)2S2O8 (3 equiv, 46.1 mmol) were added. This was stirred at 75⁰C for 30 minutes in the absence of oxygen under argon gas. DMF was evaporated off and the crude product was extracted three times with dichloromethane. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated. The product was purified by column chromatography (25% ethyl acetate/hexanes) producing compound 2 (55% yield). Mass spectroscopy confirmed the presence of a major peak at 244 m/z (compound 2).
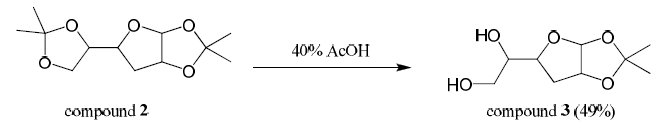
Compound 2 (8.45 mmol) was stirred in 40% acetic acid (5 mL) in a round bottom flask at room temperature for 10 hours. The reaction was quenched with saturated sodium bicarbonate, transferred to a separatory funnel where it was extracted five times with 1:1 chloroform-isopropanol. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated. The product was purified by column chromatography (100% ethyl acetate) producing compound 3 (49% yield). Mass spectroscopy confirmed the presence of a major peak at 204 m/z (compound 3).
![]()
Compound 3 (1.0 equiv, 4.14 mmol) was dissolved in dichloromethane (25 mL) and a small amount of H2O in a round bottom flask. Sodium periodate (4.5 equiv, 18.63 mmol) and sodium bicarbonate (0.2 equiv, 0.83 mmol) were added. This was stirred at room temperature overnight. The crude product was extracted three times with dichloromethane. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated producing compound 4 (85% yield). Mass spectroscopy confirmed the presence of a major peak at 172 m/z (compound 4).
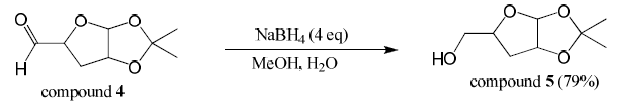
Compound 4 (1.0 equiv, 3.52 mmol) was dissolved in 90% methanol (20 mL) in a round bottom flask. Sodium borohydride (4.0 equiv, 14.1 mmol) was added. This was stirred at room temperature overnight. Methanol was evaporated off and the crude product was extracted three times with 1:1 chloroformisopropanol. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated producing compound 5 (79% yield). Mass spectroscopy confirmed the presence of a major peak at 174 m/z (compound 5).
![]()
Compound 5 (1.0 equiv, 2.85 mmol) was dissolved in dry dichloromethane (10 mL) in a dry round bottom flask. 2.5 equivalents of triethylamine (7.13 mmol) was added. The flask was cooled to 0⁰C and methanesulfonyl chloride (1.5 equiv, 4.28 mmol) was added dropwise. This was stirred at 0⁰C for 5 minutes and then for 15 minutes at room temperature. The crude product was extracted three times with dichloromethane. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated producing compound 6 (81% yield). Mass spectroscopy confirmed the presence of a major peak at 252 m/z (compound 6).
![]()
Compound 6 (1.0 equiv, 2.31 mmol) was dissolved in dry dimethylformamide (15 mL) in a dry round bottom flask. 4.0 equivalents of sodium azide (9.24 mmol) was added. The flask was stirred at 80⁰C overnight. The crude product was extracted three times with dichloromethane. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated producing compound 7 (78% yield). Mass spectroscopy confirmed the presence of a major peak at 199 m/z (compound 7).
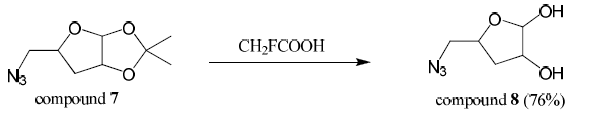
Compound 7 (1.8 mmol) was stirred with trifluoroacetic acid (3 mL) for 10 hours. Copious amounts of 1:1 methanol-toluene was added and evaporated off. This was performed four times. The crude product was extracted three times with 1:1 chloroform-isopropanol. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated producing compound 8 (76% yield). Mass spectroscopy confirmed the presence of a major peak at 159 m/z (compound 8).
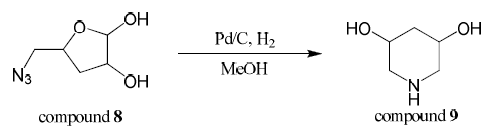
Compound 8 (1.3 mmol) was dissolved in dry methanol (3.5 mL) and powder Pd/C catalyst was added. The reaction was stirred and hydrogen gas was applied (balloon) for 24 hours. Pd/C catalyst was removed by filtering the reaction mixture through a microfilter and then methanol was evaporated off.
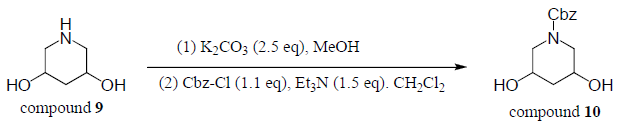
Compound 9 (1.0 equiv) was dissolved in dry methanol and 2.5 equivalents of potassium carbonate was added. The reaction was stirred at room temperature for 30 minutes. This was extracted three times with dichloromethane. The combined organic layers were dried over magnesium sulfate, filtered, and concentrated. The crude product (1.0 equiv) was then dissolved in dry dichloromethane and cooled to 0⁰C. 1.1equivalent of benzyl chloroformate and 1.5 equivalents of triethylamine were added. The reaction mixture was stirred for 1 hour at 0⁰C and then stirred for 4 hour at room temperature. The reaction did not proceed and this was confirmed with TLC.
Results and Discussion
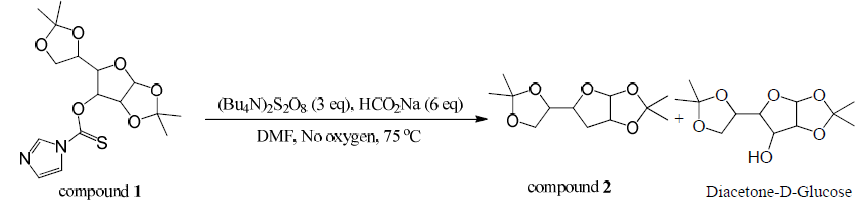
Compound 2 was isolated as well as unidentified byproducts and diacetone-D-glucose. Compound 1 was hydrolyzed and converted back to diacetone-D-glucose due to the presence of water. Despite five attempts and much effort to remove water that may have been present, a significant amount of starting material was converted back to diacetone-D-glucose. The presence of byproducts was confirmed with TLC.
![]()
We tried to selectively deprotect diol at C-1, but halfway through the reaction, the diol on compound 3 also started to get deprotected. At the end of the reaction, a mixture of compound 3 and double diol deprotection byproduct were observed.
Mass spectroscopy showed several peaks that were above the expected mass of compound 9 and compound 8. From this result we concluded that polymerization occurred. Even though we found we did not obtain compound 9 due to polymerization, we tried to run the last step (from compound 9 to compound 10) presented in the experimental methods section. As expected, we did not obtain compound 10. This result was confirmed by mass spectroscopy.
Other steps besides those three steps mentioned above proceeded as expected. The percent yield of each step is shown above in the experimental section. The presence of the product of each step was confirmed with mass spectroscopy. Also, 1HNMR was used to confirm the production of compound 5 and 7.4, 5
We found that the method presented above to convert diacetone-D-glucose to 2-DOS mimic was not efficient. We lost a significant amount of material in the second step (compound 1 to compound 2) due to unexpected hydrolysis and the third step (compound 2 to compound 3) due to double diol deprotection. In addition, we could not obtain compound 9 because of polymerization. Because this method requires too many steps it is difficult to get a good amount of final product. Due to these reasons we decided to give up this method and try a new method, which takes fewer steps, to synthesize 2-Dos mimic.
Conclusion
The method we had used to synthesize 2-Dos mimic from diacetone-D-glucose was given up due to problems that occurred in step 2, 3, and 9; conversion of compound 2 back to diacetone-D-glucose and production of unexpected byproducts in step 2, production of byproduct by double diol deprotection in step 3, and polymerization occurring in step 9. A new method, which uses xylitol as the starting material, is to be used to synthesize 2-DOS mimic because by the new method, we can avoid the problems in the previous method and the new method takes fewer steps.
Results
- Malim, M.; Hauber, J.; Le S.; Maizel, J.; Cullen B. Trans-dominant inactivation of HTLV-I and HIV-
1 gene expression by mutation of the HTLV-I Rex transactivator. Nature 1989, 338, 254-257. - Hobbie, S. N.; Pfister, P.; Brull, C.; Westhof, E.; Bottger, D. C. Analysis of the contribution of
individual substituents in 4,6-aminoglycoside-ribosome interaction. Antimicrob. Agents Chemother
2005, 49, 5112-5118. - Frain, D.; Kirby, F.; McArdle, P.; O’Leary, P. The Synthesis of AraBOX, a New 4,4’-Bis(oxazoline),
from Novel Pentitol-Derived Bis-beta-amino Alcohols. Synlett 2009, 8, 1261-1264 - Compound 5
1HNMR (300MHz, CDCl3): δH= 1.30 (6H, s), 1.94 (2H, s), 3.66 (2H, d), 3.83 (1H, m), 4.22 (1H, q),
5.26 (aH, d). - Compound 7
1HNMR (300MHz, CDCl3): δH= 1.27 (6H, s), 1.51 (2H, m), 1.94 (2H, m), 3.81 (1H, m), 4.21 (1H,
q), 5.24 (1H, d).