
Spring Cullen and Mentor: Dr. Mark Clement, Computer Science: Bioinformatics
Project Purpose
Epigenetics is of vital importance, comparable to genetics, in predicting the outcome of illnesses. Bisulfite sequencing (BS) provides short DNA fragments that must be mapped in order to discover epigenetic markers. The purpose of this project is to find an optimal cost matrix to map sequences from BS DNA to a reference genome to determine patterns of methylation. A more efficient BS cost matrix will contribute to the ongoing effort to understand the epigenome and analyze data collected.
Project Importance
This project revolves around deepening the scientific understanding of epigenetics–the study of changes in organisms due to alterations regarding gene expression. These heritable changes in gene expression do not involve changes to the DNA sequence. Normal DNA is made up of strands of T, A, C and G nucleotides. DNA methylation is a process where a methyl group is added to a position along a DNA strand. These added methyl groups can cause 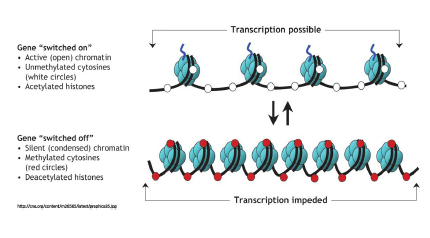 a normal DNA strand to bind so tightly that the genetic information cannot be accessed or transcribed —literally turning genes “off” or “on”. The epigenome consists of all the methyl-group tags and other compounds that have been added to a DNA strand. External sources such as diet, pollution and traumatic experiences can affect the epigenome—altering gene expression. This allows us to study disease beyond DNA mutations. Studies by Fienberg and Vogelstein in 1983 found that genes of cancer cells were substantially hypomethylated when compared to normal tissue. If epigenetic data could be correctly matched and tracked, many diseases could be circumvented. For example, if the cause of aberrant methylation patterns, common in cancers, could be traced and understood then the risk of cancer could be decreased.
a normal DNA strand to bind so tightly that the genetic information cannot be accessed or transcribed —literally turning genes “off” or “on”. The epigenome consists of all the methyl-group tags and other compounds that have been added to a DNA strand. External sources such as diet, pollution and traumatic experiences can affect the epigenome—altering gene expression. This allows us to study disease beyond DNA mutations. Studies by Fienberg and Vogelstein in 1983 found that genes of cancer cells were substantially hypomethylated when compared to normal tissue. If epigenetic data could be correctly matched and tracked, many diseases could be circumvented. For example, if the cause of aberrant methylation patterns, common in cancers, could be traced and understood then the risk of cancer could be decreased.
It is impossible to correctly map methylation through previously used sequencing techniques. Epigenetic events are found by bisulfite sequencing. Treatment of DNA with bisulfite converts C residues to U and when the bisulfite treated DNA is sequenced, the U will be seen as a T. The mapping algorithm must then differentiate between real C-T mutations and those due to methylation.
It is very difficult to read methylated DNA sequences and analyze results due to the possible error sources. Mapping involves matching DNA sequence fragments to a reference genome. During read mapping, one DNA sequence is aligned to another to see how similar they are. A cost matrix is used in the alignment process to determine a score for the DNA fragment. This score is based on how similar the matched sequence is. Different cost matrices generate different mappings by giving varying scores to DNA fragments, changing results.
Methodology
By using the program Art_Illumina I was able to create a synthetic data set to use as a simulation study under different matrices. The parameters used for the single-end simulation : read length of 150, genome masking ‘N’ cutoff frequency of 1 in 150, a fold coverage of 10X and a combined profile type. a random seed was generated for every run. The output from the simulation was saved in a .sam file. GNUMAP was then used on these generated reads using the following parameters: Verbose 1, an align percentage of 90%, 1 thread, Mer size 10, Using jump size of 5, using a gap score of -1 and accounting for a maximum gaps of 3. The .sam file from art_illumina was converted into a vector with an average hash size of 1048576. Data collected from GNUMAP runs consisted of Total Run Time, Sequences matched, Sequences not matched and output written to a second .sam file.
Each Illumina synthetic data set was run with GNUMAP in combinated with a error cost matrix. These matrices are used to align a set of reads, or short DNA sequences, with the reference genome. According to the GNUMAP documentation, the current alignment score matrix used is described as 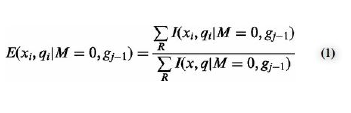 follows: “The position-weight matrix used by the algorithm as default is a simple matrix with (-1) as the mismatch score and (+1) as the match score.” This very basic scoring matrix does not take into account the errors appear to occur when DNA undergoes bisulfite sequencing. The following is a formula used by researchers at Nucleic Acids Research to determine the average percent of methylated cytosines in a human genome.[1] Data acquired by this formula I was able to predict the probability of read being methylated in an average genome. From there two alternative cost matrices were created. Acting as a control, one result in no penalty when a C was matched to T. The second accounted for the average amount of methylatation, namedly 18.1375% of a genome.
follows: “The position-weight matrix used by the algorithm as default is a simple matrix with (-1) as the mismatch score and (+1) as the match score.” This very basic scoring matrix does not take into account the errors appear to occur when DNA undergoes bisulfite sequencing. The following is a formula used by researchers at Nucleic Acids Research to determine the average percent of methylated cytosines in a human genome.[1] Data acquired by this formula I was able to predict the probability of read being methylated in an average genome. From there two alternative cost matrices were created. Acting as a control, one result in no penalty when a C was matched to T. The second accounted for the average amount of methylatation, namedly 18.1375% of a genome.
Results
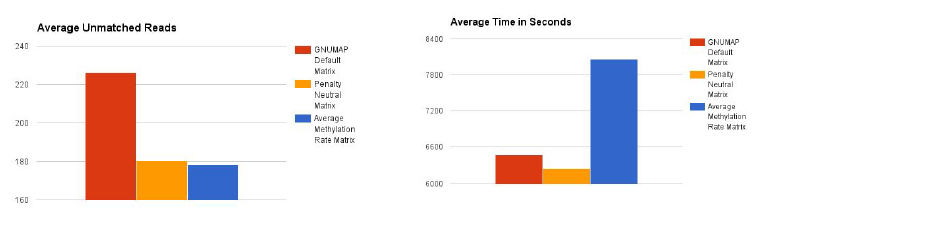
The average runtime the methylation focused matrix was 8066 seconds, while the default averaged at 6460s and the penalty neutral matrix at 6255s. In terms of average unmatched reads the GNUMAP default left 226 reads unmatched, while the penalty neutral matrix averaged 180 unmatched reads while the average methylation matrix averaged at 178 unmatched reads.
Discussion and Conclusion
Results revealed that in order for read mapping to be most accurate to handle bisulfite treated DNA sequences, the error matrices must conditioned to better replicate real world methylation on reads but that increased accuracy comes at a price, the time it takes to sequence the genome. Thus the tradeoff must be considered when using GNUMAP matrices to map reads to a reference genome. More research should be done to determine specific nucleic patterns surrounding regions of methylation in order to increase accuracy. It would be best continue research in this field to develop more specialized and accurate mapping and synthetic simulation of DNA that has undergone bisulfite treatment.