
Elise Wilson and Faculty Mentor: John C. Price, Department of Chemistry and Biochemistry
Introduction
Mass spectrometry utilizes ionized atoms or molecules, and separates these particles according to their mass-to-charge (m/z) ratios. Mass spectrometry is a commonly used analytical tool in physics, chemistry, biochemistry, and pharmaceutical science.1 In the biochemical study of protein kinetics, mass spectrometry can be used to detect protein turnover in vivo by metabolically labeling tissue with deuterium (or hydrogen with a proton and a neutron in the nucleus) and detecting the change in the m/z over time.2,3 As the supplied deuterium is used in the synthesis of new protein over the course of several days, the relative abundance of heavier m/z ratios increases and this change of abundances can be used to calculate the percent new peptide over a period of time.4
In theory, mass defects can also be used to measure protein turnover. The mass defect is the difference between an isotope’s exact mass and its calculated, or nominal mass. When the mass of an isotope is calculated, the individual masses of the protons, neutrons, and electrons that make up the isotope are added. But this mass is different from the exact mass due to the binding energy according to the relation E=mc2, where E is the binding energy, m is the mass change, and c is the speed of light.5 This mass defect can be measured using a mass spectrometer with a high mass accuracy. The incorporation of heavy isotopes into newly synthesized proteins causes the protein itself to have a slightly different experimentally observed mass than the expected mass; detectable as a m/z shift, as seen in Figure 1. The theoretical mass defect over time can then be used to calculate the protein turnover.
Methodology
Mice were metabolically labeled with deuterium by providing the mice with a constant supply of deuterated drinking water. This heavy labels the amino acids due to rapid water equilibrium throughout the body. As previously demonstrated, these heavy amino acids are then incorporated into the newly synthesized proteins and the rate of incorporation can be measured by mass spectrometry.3
Mass spectrometry shows that each peptide has a unique isotopic distribution of masses due to the natural distribution of isotopes. This isotope distribution shifts over time as heavy amino acids are incorporated into newly synthesized proteins. This change in the isotope pattern can be used to calculate the relative change in isotopic intensity (ΔIx) (see Figure 1). Then, the percent new peptide can be calculated as the ratio of ΔIx to the total theoretical change expected at available isotopic enrichment (ΔIx*) so that the Percent New Peptide = ΔIx/ΔIx*. The percent new peptide can then be plotted against time and the rate of protein synthesis can then be calculated by using a best fit curve.
To calculate the percent new peptide from the mass defect we first measure the isotope pattern at each time point, and then calculate the m/z difference from M0 (ΔDx) (see Figure 1). We then calculate the mass defect (T) by subtracting the theoretical m/z difference from M0 (ΔDx*) so that T = ΔDx − ΔDx*. Finally, the percent new peptide can be calculated by dividing T by the mass defect expected at the available isotopic enrichment (T*) so that the Percent New Peptide = T/T*. Again, the rate of protein synthesis can be calculated by using a best fit curve like with the abundance intensity calculations.
Results and Discussion
Orbitrap vs Quadrupole-Time-of-Flight
Accurate mass measurements are necessary to sucessfully calculate protein sythesis rates from the mass defect and the type of mass spectrometer used to collect the data can significantly impact the mass accuracy of those measurments. We explored the mass defect measurements and calculations on two different mass spectrometers, Orbitrap and Quadrupole-Time-of-Flight (QTOF), to optimize the use of mass defect for protein turnover measurements on each. We found that, as expected, the higher resolution capabilities of the Orbitrap instrument generally resulted in less deviation (at 2/3 the deviation of the QTOF) from the peptide’s theoretical m/z for the time zero measurements (unlabeled sample). Interestingly, the 20K resolution QTOF measurements had a 1.5 times greater deviation from the expected m/z than the 10K resolution measurements, suggesting that the QTOF performs more accurately at the lower resolution. Also, while the deviation for the M0 measurement was comparable across all the different instruments and resolutions, the M2 and M3 measurements taken on the Orbitrap showed negative deviation from the expected m/z. Another point to consider is that the deviation within the isotopic envelope is more consistent for the QTOF measurements than for the Orbitrap measurements. Therefore, if greater accuracy at time zero is required, then the higher resolutions on the orbitrap would be the better choice while if consistent error within the isotopic envelope is more important, then the measurements would be better performed on the QTOF.
The rates of peptide synthesis were calculated by fitting to a logarithmic curve. No parameters were used to force the curve to zero, accommodating for negative percent new peptide values obtained from the mass defect percent new peptide calculations. Except for the 20K resolution QTOF data, the intensity curves gave similar rates. The QTOF 10K and Orbitrap 480K mass defect curves also resulted in similar rates. While not agreeing with the other data points, the QTOF 20K gave similar results for both the intensity and the mass defect. The higher resolution measurements seem to have a greater agreement between the intensity rates and mass defect rates, though they don’t necessarily match up to rates calculated from the other measurements.
Conclusions
This method is a novel use of mass defect and will offer an additional way to measure the percent new peptide. The mass defect and isotope abundances of m/z can be combined for a single peptide, and using both methods together would provide seven separate measurements per peptide to calculate the rate of protein turnover. The additional data points will greatly improve statistical confidence in the results. Additionally, protein kinetic studies can now be performed on several different kinds of mass spectrometers by using the mass defect, whereas previous isotope abundance studies were most reliable on time-of-flight mass spectrometers. This exciting new method for determining the rate of protein synthesis will be a useful tool for the analysis of protein kinetics. The Price Lab will continue to fine tune the calculations and instrument parameters for using the mass defect to make rate measurements as well as develop software to automate this process.
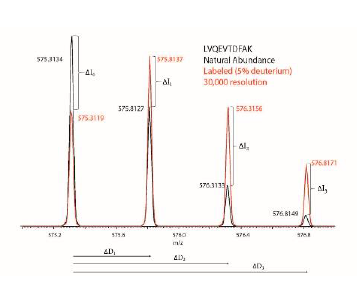 Figure 1. The change in the isotopic envelope from the natural abundance to maximum (5%) deuterium labeling. The change in abundance over time is represented by ΔIx while the mass defect can be calculated using the m/z difference from M0 (the peak with the lowest m/z in the isotopic envelope), represented by ΔDx.
Figure 1. The change in the isotopic envelope from the natural abundance to maximum (5%) deuterium labeling. The change in abundance over time is represented by ΔIx while the mass defect can be calculated using the m/z difference from M0 (the peak with the lowest m/z in the isotopic envelope), represented by ΔDx.
1 Dass, C., Basics of Mass Spectrometry. In Fundamentals of Contemporary Mass Spectrometry, John Wiley & Sons, Inc.: 2006; pp 1-14.
2 Guan, S.; Price, J. C.; Ghaemmaghaml, S.; Prusiner, S. B.; Burlingame, A. L. Compartment modeling for mammalian protein turnover studies by stable isotope metabolic labeling. Anal. Chem. 2012, 84, 4014-4021.
3 Louie, K. B.; Bowen, B. P.; McAlhany, S.; Huang, Y.; Price, J. C.; Mao, J.-h.; Hellerstein, M.; Northen, T. R. Mass spectrometry imaging for in situ kinetic histochemistry. Sci. Rep. 2013, 3.
4Price, J. C.; Khambatta, C. F.; Li, K. W.; Bruss, M. D.; Shankaran, M.; Dalidd, M.; Floreani, N. A.; Roberts, L. S.; Turner, S. M.; Holmes, W. E.; Hellerstein, M. K. The Effect of Long Term Calorie Restriction on in Vivo Hepatic Proteostatis: A Novel Combination of Dynamic and Quantitative Proteomics. Mol. Cell. Proteomics 2012, 11, 1801-1814.
5 Sleno, L. The use of mass defect in modern mass spectrometry. J. Mass. Spectrom. 2012, 47, 226-236.