
Michael Porter and Dr. John C. Price, Chemistry and Biochemistry
Introduction
Proteins are involved in nearly every cellular process. They transport molecules, replicate DNA, catalyze reactions, respond to stimuli, and form the structural framework of the cell. The diversity of protein function has led to an interest in understanding the cellular production and control of individual proteins which in turn is key to understanding how the cell responds to the environment and what occurs on a molecular level during disease.
While mass spectrometry tells us what proteins are present, historically it tells us nothing of the rate at which they are formed and degraded. The rate of protein formation and degradation, also known as turnover, is important in understanding how cellular processes interact and are regulated. This technique relies on stableisotope labeling which allows mass spectrometry to differentiate newly synthesized protein from old. Isotopes are a chemical variant of an element, like carbon, which contain one or more extra neutrons. Isotopes are naturally occurring and create characteristic “isotopic envelopes” in mass spectra. By enriching the amount of heavy isotopes in the body, we observe that isotope abundances change as the amount of newly synthesized molecules in a population increase. The rate of turnover can then be calculated by using the shifts observed at different time points after adding a heavy isotope.
Current methods for calculating protein turnover rely on peak heights which represent the abundance of a certain isotope. The accuracy of this method is affected by the noise in the spectrum and for low abundance signals cannot always produce an accurate result. Recent advances in high resolution mass spectrometry have produced instruments so sensitive to measuring mass differences that they are able to detect differences due to Einstein’s equation E=mc2. These differences, called the mass defect, are caused by the release of energy when two atoms form a bond and are related to the isotopic makeup of the protein. The mass defect can be used along with the peak heights to provide more accurate measurements of turnover rate.
The current software tools for this kind of analysis are incomplete and require tedious work by hand which introduces random error into the results.
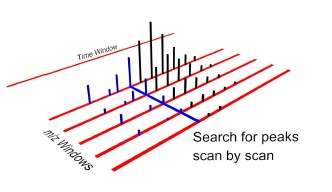
Figure 1. As a sliding window is moved through time, any peaks falling within the m/z windows are extracted.
Methodology
To extract the isotopic envelopes, a method was developed which employs vector angles. Rather than extract the peaks individually as is done in other approaches, our method looks for the entire isotopic envelope and tracks it throughout time.
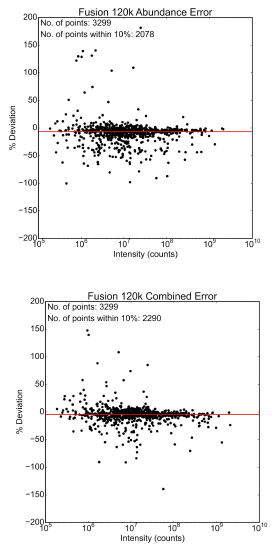
Figure 2. Accuracy of protein turnover rates calculated using only abundance (top) or a combination of both abundance and mass defect (bottom). The combination of both methods increases the accuracy of the rates.
Results
Our approach is capable of extracting more information than the vendor specific software currently in use. Our approach also led to an increase in overall accuracy by combining the mass defect measurements with the abundance measurements to calculate the turnover rate (Figure 2).
Discussion
Calculating protein turnover rates has been limited to only those instruments which measure isotope abundances with a very high accuracy. By utilizing the mass defect, we have been able to show an increase in the accuracy of our measurements of protein turnover. Our vector based approach has proven capable of accurately extracting isotopic envelopes from complex datasets and works with data from any instrument manufacturer.
Conclusion
The software we developed is crossplatform and crossinstrumental which allows any lab with a mass spectrometer to carry out protein turnover experiments. The methods we developed facilitate collaboration between labs and is easy to adapt to other uses other than turnover calculations.