
Bradley Hanks and Dr. Brian Jensen, Mechanical Engineering
Project Introduction
Non-viral gene therapy is an area of study which includes various techniques to incorporate foreign molecules into the DNA of a cell. The ultimate goal of gene therapy is to replace mutated or damaged genes with healing genes.
Nanoinjection is a method of non-viral gene therapy developed at BYU which utilizes an electrically charged lance to attract and deliver DNA into the cell1. The lance is charged in order to attract DNA to itself and then lowered into the cell culture. The lances pierce holes, or pores, passing the DNA into the cytoplasm and potentially the nucleus. A brief summary of this process is shown in Figure 1. Current development is focused on enhanced expression of DNA delivered to an entire culture of cells from an array of lances. Nanoinjection research has the potential to transform the field of gene therapy by offering a transfection technique with both high efficiency and viability.
Two aspects of nanoinjection were to be studied: the effects of the growth phase of the cell and the localization and quantification of DNA delivery. Other methods of gene therapy have identified that optimal transfection occurs during the division of the cell as the nuclear membrane dissolves2,3. A recent study from UC Berkeley was published in December of 2014 which confirmed the hypothesis that transfection efficiency increased during cell division4. Cells that were synchronized to the G2/M phase displayed the highest transfection rates. These results allowed the focus of this study to be narrowed to the localization and quantification of DNA delivery.
The ability to track or localize and quantify the amount of DNA is helpful in understanding how to increase the efficiency of the delivery. A radioactive tag was included on the DNA delivered to the cells which was used to track and approximate the number of DNA strands within the cell. A second process called enucleation separated the cell into its sub components to determine where the DNA strands were located within the cell. It was hypothesized that a combination of the radioactive DNA and enucleation would enable localization and quantification of the average amount of DNA delivered to a culture of cells post nanoinjection.
Methodology
The project consisted of four processes: radioactive tagging, nanoinjection, enucleation, and scintillation. A DNA plasmid was linearized and tagged with a radioactive label. A culture of HeLa cells was nanoinjected with the radioactive DNA strand. The injection solution, the cytoplasmic content, and the nuclei were separated through enucleation. A liquid scintillator counted the samples in order to localize and quantify the average amount of DNA in the cells. A detailed description of each portion of the experiment is included below.
Radioactive Tagging – pCAG-GFP plasmid was linearized using HindIII, a restriction enzyme which severs the plasmid to expose several nucleotides on the DNA strand. Free nucleotides (dATP, dCTP, dGTP, and dTTP) were added and attached to the exposed nucleotides. In addition to standard dTTP, dTTP labeled with α-32P was added in solution. This resulted in a solution of linearized plasmid, part of which was tagged with a radioactive label.
Nanoinjection – HeLa cells were cultured in four 6-well plates. The wells were categorized into three groups: negative controls, positive controls, and treatment wells. The negative control received no DNA and was not injected. The positive control received DNA but was not injected. The treatment wells received DNA and were injected. 470 ng of linearized, radiotagged DNA were added to the non-negative wells prior to injection. An automated nanoinjection system5 was used with various electrical and timing settings described in the table below.
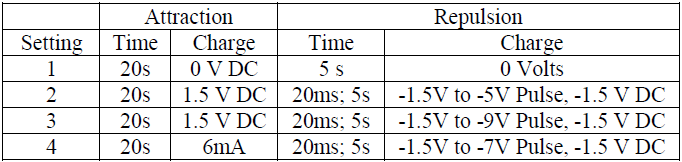
The injection step was divided into two phases: attraction, and repulsion. During attraction, a charge was applied across the lance array for 20 seconds to attract the DNA to the lances. The lance array was then lowered to create pores in the cell membrane. Ten pulses followed by a repulsion charge were applied to release the DNA from the lances into the cells.
Enucleation – The enucleation process was performed using an Active Motif Nuclear Extract kit. The injection solution was removed from each well and placed in 24 scintillation vials. The cells were suspended in a hypotonic buffer, detergent was added, and the cells were vortexed to rupture the cell membranes. The ruptured cells were centrifuged again to separate the nuclei from other cytoplasmic content. The supernatant, or other cytoplasmic content, was removed and placed in a second set of 24 scintillation vials. The nuclear pellets were then resuspended in PBS and placed in a third set of 24 scintillation vials.
Scintillation – A liquid scintillator analyzer (Tri-Carb B2910TR, PerkinElmer) was used to measure radioactive emission from the tagged DNA in the cell samples. The excess injection fluid, cytoplasmic content, and nuclear pellet samples were analyzed to determine the radioactive emission present in each. The data was correlated to an approximate number of strands of DNA per vial with a dilution calibration.
Results
The experiments were initially run using settings 1, 2, and 3 from Table 1. After these initial experiments, it became evident that there was significant cell loss during the procedure and a cell count during the experiment was necessary for an accurate approximation of DNA strands per cell. The number of DNA strands detectible was also found to be too high to determine the amount of DNA in the nucleus. The final data set, with setting 4, includes a more accurate cell count and increased resolution. Figures 2 and 3 show the average number of DNA strands in the cytoplasm and nucleus, respectively, for each of the 4 electrical settings. Error bars are shown as one standard deviation. Horizontal bars are also shown to denote statistical significance. Figure 2 shows that there was an average of 50,000 strands of DNA/cell delivered to the cytoplasm. Figure 3 shows that there was an average of 1350 strands of DNA/cell delivered to the nuclei.
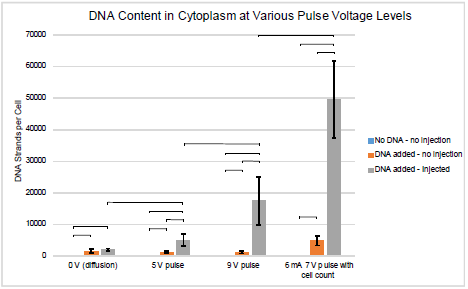
Figure 2. DNA present in cytoplasm
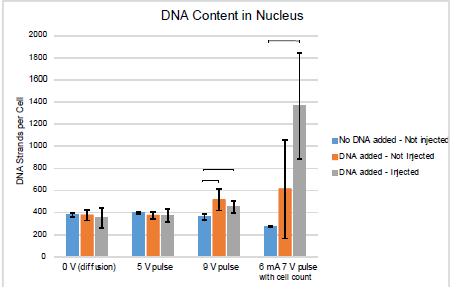
Figure 3. DNA present in nucleus
Conclusions
The results have shown that nanoinjection is a viable option for introducing genes into the cytoplasm and likely the nucleus as well. Figure 2 shows that as the electrical environment increases with intensity, the amount of DNA present increases as well. With no electrical environment present (setting 1), no difference can be seen between the positive and treated samples. Increasing the peak pulse from -5V to -9V shows a direct increase in the amount of DNA present. While setting 4 may not be compared as directly with settings 2 and 3, the improved resolution and cell count provides greater confidence in the accuracy of these results.
No statistical significance can be drawn from the nucleic contents shown in Figure 3. Drastic improvements were seen in the resolution when the concentration of the radiotagged DNA was enhanced. It is hypothesized that with further testing the standard deviation of samples could be reduced to show statistical significance between positive and treated samples.
Discussion
Nanoinjection has proven to be a method of introducing foreign material into the cell with great cell viability. While this study did not track viability throughout the experiment, it provided great insight into the location of the delivery of the genetic material. Further studies may be performed in order to attempt greater understanding of the content of the nuclei. Two areas of development in order to further this study would be an improved method of cell counts prior to scintillation. A second area to improve the results of the study would be an increased resolution on the number of strands of DNA. For much of the experiment, the amount of DNA present in the nucleus was too close to or below the minimum amount.
Special thanks to Nick Gregory and Nelson Warner who helped in preparing, performing, and analyzing experiments.
Sources
- Lindstrom, Z. K.; Brewer, S. J.; Easter, M. A.; Jensen, B. D.; Burnett, S. H., “Injections of Propidium Iodide into HeLa Culture Cells Using a Nanoinjection Lance Array,” ASME 2013 International Design Engineering Technical Conference & Computers and Information in Engineering Conference, Portland, 2013.
- Pichardo, S.; Togtema, M.; Jackson, R.; Zehbe, I.; Curiel, L., “Influence of cell line and cell cycle phase on sonoporation transfection efficiency in cervical carcinoma cells under the same physical conditions,” Ultrasonics, Ferroelectrics, and Frequency Control, IEEE Transactions on, vol.60, no.2, pp.432,435, February 2013.
- Yu, Jian-Ning; Ma, Suo-Feng; Miao, De-Qiang; Tan, Xiu-Wen; Liu, Xin-Yong; Lu, Jin-Hua; Tan, Jing-He, “Effects of Cell Cycle Status on the Efficiency of Liposome-mediated Gene Transfection in Mouse Fetal Fibroblasts,” Journal of Reproduction and Development, vol.52, no.3, pp.373,382, June 2006.
- Lin, Steven; Staahl, Brett; Alla, Ravi K.; Doudna, Jennifer A., “Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery,” eLife 2014; 10.7554/eLife.04766
- Lindstrom, Zachary K.; Gregory, Nicholas; Jensen, Brian D., “Design and testing of an automated multi-cell nanoinjection system,” 19th Design for Manufacturing and the Life Cycle Conference, vol. 4, August 2014