
Ryan Hyatt and Joshua L. Andersen, Department of Chemistry and Biochemistry
Introduction
Autophagy is an adaptive catabolic process of self-digestion, a process by which the cell recycles aged organelles and other structures. Interestingly, changes in the regulation of autophagy have been linked to infections, cancers, neurodegeneration, aging, and heart disease (Arroyo, Daniela S. et al. 2014). Understanding the mechanisms which control autophagy will aid in better treatment of those diseases, specifically in the development of effective pharmaceuticals.
ATG9 is a multimembrane-spanning protein required for induction of autophagy. Mack et al. 2012 published a paper providing evidence that ATG9 contributes membranous material to the growing autophagosomes (Mack et al., 2012) and may participate in the trafficking of autophagosomes throughout the cell.
Recently, the Andersen lab at BYU found that in order for ATG9 to actively contribute to growing autophagasomes, it must be phosphorylated at S761 and bind to 14-3-3ζ (see figure 1).
We hypothesize that ULK1 acts as a scaffold to bring active AMPK and ATG9a to phosphorylate the S761 motif (of ATG9) under normal conditions, thus activating ATG9.
In order to test this hypotheses, I have conducted the following mutagenesis to determine the binding regions between ULK1 in nutrient replete, normoxia conditions: deletion of the N terminus of ULK1 (Amino Acids 71-249), deletion of the Serine/Threonine rich region of ULK1 (Amino Acids 299-621) and the C terminus of ULK1 (Amino Acids 849-1071). Preliminary data suggests that ULK1’s interaction with ATG9 takes place at the N terminus of ATG9, depending on kinase activity of ULK1. These findings are supported by Chan et al. which revealed that a 7 amino acid motif on the C terminus of ULK1 is critical for ULK1 activation via AMPK.
Methodology
Site Directed Mutagenesis. The ATG9 and ULK1 mutant plasmids were developed using the Q5 Site-Directed Mutagenesis kit from New England Biolabs (catalog number E0554S) according to the manufacturer’s protocol.
Plasmids. The HA-Atg9A plasmid was kindly provided by Sharon Tooze, London Research Institute, United Kingdom. The HA-ULK1 plasmid was purchased from Addgene (catalog number 31963).
Antibodies. Antibodies to Hemagluttinin (HA), ULK1, Actin and phosphorylated 14-3-3ζ motifs were obtained from Cell Signaling, Inc.
Results
Unfortunately, our preliminary immunoprecipitation and immunoblotting with these mutants have proven inconclusive, so we have begun the process of generating new, more extensive deletion mutants at the following regions (N-terminus amino acids 1-249), deletion of amino acids 250-299 of the Serine/Threonine rich region, deletion of amino acids 622-849 at the C terminus, and deletion of the specific 7 amino acid motif in the C terminus, in conjunction with the findings of Chan et al. 2009. The rational for these deletion regions comes from our understanding of the location of motifs that are believed to be necessary for protein folding (see figure 2), which we want to preserve. It is anticipated that by evaluating the interactions of these mutants with ATG9, we can pinpoint which amino acids in which region are necessary for ULK1 – ATG9 mediated control of autophagy in normoxia and nutrient replete conditions.
Discussion
Determining the Amino Acids critical for interaction between ULK1 and ATG9 would further elucidate the complexity and details of the regulation of Autophagy in basal conditions. After that interaction is understood, we aim to broaden our study by attempting to understand the co-interaction with AMPK. With this knowledge, we hope to develop a better understanding of the mechanisms that contribute to the abnormal regulation of ATG9, especially in the aforementioned diseases.
Conclusion
Given the necessity for ULK1 to be phosphorylated at the C terminus in order for it to activate ATG9’s activity in Autophagy (Chan et al. 2009), we speculate that there is a critical interaction between the C terminus of ULK1 and the N terminus of ATG9. We anticipate that this will be confirmed in the very near future.
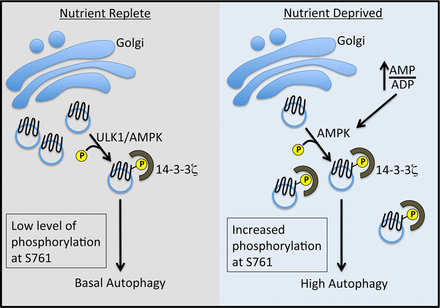
Figure 1. Schematic diagram of the steps of autophagy. Weerasekara et. Al. 2014, Metabolic-Stress-Induced Rearrangement of the 14-3-3ζ Interactome Promotes Autophagy via a ULK1- and AMPK-Regulated 14-3-3ζ Interaction with Phosphorylated Atg9, Mol. Cell. Biol. December 2014 vol. 34 no. 24
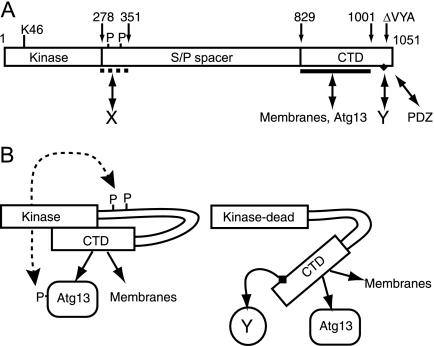
Figure 2. Chan, et al. “Kinase-Inactivated ULK Proteins Inhibit Autophagy via Their Conserved C-Terminal Domains Using an Atg13-Independent Mechanism.” Molecular and Cellular Biology 29.1 (2009): 157–171. PMC. Web. 21 Mar. 2015.
References
- Arroyo, Daniela S. et al. 2014. “Autophagy in Inflammation, Infection, Neurodegeneration and Cancer.” International immunopharmacology 18.1 (2014): 55–65. PMC. Web. 14 Apr. 2015.
- Mack et al. 2012. “AMPK-Dependent Phosphorylation of ULK1 Regulates ATG9 Localization.” Autophagy 8.8 (2012): 1197–1214. PMC. Web. 21 Mar. 2015.
- Weerasekara et al. 2014. Metabolic-Stress-Induced Rearrangement of the 14-3-3ζ Interactome Promotes Autophagy via a ULK1- and AMPK-Regulated 14-3-3ζ Interaction with Phosphorylated Atg9, Mol. Cell. Biol. December 2014 vol. 34 no. 24 4379-4388
- Chan et al. 2009. “Kinase-Inactivated ULK Proteins Inhibit Autophagy via Their Conserved C-Terminal Domains Using an Atg13-Independent Mechanism.” Molecular and Cellular Biology 29.1 (2009): 157–171. PMC. Web. 21 Mar. 2015.