
Michaela Squire and Dr. Scott Steffensen, Psychology
Introduction
With today’s society’s alcohol abuse, there has been a large economic burden of several hundred billion dollars annually. In addition, 80% of those who are dependent on alcohol are also smokers, and the use of nicotine has remained relatively high in those who use alcohol, displaying the co-dependence of nicotine and alcohol. Due to the high percentage of apparent co-dependence of nicotine and ethanol (EtOH), it has been theorized that the addictive nature of these drugs may act through similar mechanisms, specifically through nicotinic acetylcholine receptors (nAChRs) on presynaptic gamma-aminobutyric acid (GABA) neurons. The purpose of this project was to evaluate this possible involvement of the α6 subunits of the nAChRs on modulating the inhibitory effects of EtOH through dopamine (DA) release from GABAergic neurons in the nucleus accumbens (NAc).
Methods
This project involved C57BL/6 (WT) mice between 18 and 22 days old. The DAergic reward system is well-conserved across mammalian species and so the mouse brain provides a very accurate model of the effects of EtOH on this specific neural system. The mice were anesthetized by intraperitoneal (IP) injection of ketamine (70 mg/kg), and decapitated. The brains were dissected and rapidly cooled in a low Ca2+, high Mg2+, ice cold solution. Slices (400 μm) were then obtained using a vibratome sapphire blade viewed under a microscope, while being bathed in a continuous flow of artificial cerebrospinal fluid (ACSF) held at 35.7 °C. The nicotinic cholinergic antagonists Dihydro-beta-erythroidine (DHβE) and MII conotoxin were dissolved into ACSF for superfusion onto brain slices at a given molar concentration. For Fast Scan Cyclic Voltammetry (FSCV) recordings, carbon fiber electrodes (CFE) were positioned below the surface of the slice in the NAc core. DA release was evoked every 2 min. by a stimulating electrode (Plastics One, Roanoke, VA) placed across the CFE. Stimulations were performed periodically every 2 min. and DA levels were monitored. Once the stimulated DA response was stable for five successive collections, baseline measurements were taken. Using FSCV, in the nucleus accumbens shell (NAcS) and the nucleus accumbens core (NAcC), DA release was analyzed in the presence of nicotine with both the antagonists DHβE and MII conotoxin. Finally, in order to support the existence of α6 nAChRs in the NAcC as opposed to the NAcS, we ran a dose response of both EtOH (20-160 mM) and nicotine (0.01-10 uM) in th7ese regions. We then tested the effects of DHβE and MII conotoxin and observed its effects in blocking the inhibition caused by nicotine and EtOH in both the NAcC and NAcS, displaying specifically whether the α6-nAChRs were present in either anatomical region of the NAc.
Results
Using FSCV, we evaluated the effects of nicotine (0.01 – 10 μM) on evoked DA release in the NAc shell. Nicotine significantly decreased the peak amplitude of the DA signal with an IC50 of approximately 0.1 μM (Fig. 1A,B; 0.01 μM, 0.1 μM, 10.0 μM). We evaluated the frequency 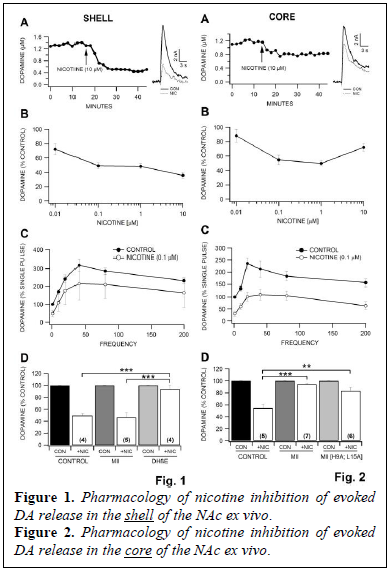 dependency for nicotine effects on DA release in the NAc shell. We applied stimulation trains (10 pulses) at varying frequencies ranging from 10 – 200 Hz and compared the response produced by multiple pulses to that obtained by a single pulse. The maximum release occurred at 40 Hz stimulation in the NAc shell (Fig. 1C). Nicotine significantly decreased the amplitude of the DA signal across all frequencies tested. Next we evaluated the effects of various nAChR antagonists on evoked DA release in the NAc shell. We analyzed evoked DA release in the presence of nicotine with the selective α6* antagonist α-CtxMII (100 nM) and also the partially selective α4β2* antagonist DHβE (50 μM). Superfusion of α-CtxMII did not block the inhibition caused by 0.1 μM nicotine in the NAc shell, however, superfusion of DHβE significantly inhibited the DA signal about 40% from baseline (Fig. 1D). Additionally, DHβE blockade of nicotine induced DA reductions was significantly increased when compared to α-CtxMII experiments. Using FSCV, we evaluated the effects of nicotine (0.01 – 10 μM) on evoked DA release in the NAc core. Nicotine significantly decreased the peak amplitude of the DA signal with an IC50 of approximately 0.1 μM (Fig. 2A,B; 0.01 μM, 0.1 μM, 1.0 μM, 10.0 μM). We noted varying results at higher concentrations of nicotine (10 μM) in the NAc shell (Fig. 1B) vs the NAc core (Fig.2B). The frequency dependency for nicotine effects on DA release in the NAc core (Fig. 2C) was similar to the shell; however, the maximum release occurred at 20 Hz rather than 40 Hz.
dependency for nicotine effects on DA release in the NAc shell. We applied stimulation trains (10 pulses) at varying frequencies ranging from 10 – 200 Hz and compared the response produced by multiple pulses to that obtained by a single pulse. The maximum release occurred at 40 Hz stimulation in the NAc shell (Fig. 1C). Nicotine significantly decreased the amplitude of the DA signal across all frequencies tested. Next we evaluated the effects of various nAChR antagonists on evoked DA release in the NAc shell. We analyzed evoked DA release in the presence of nicotine with the selective α6* antagonist α-CtxMII (100 nM) and also the partially selective α4β2* antagonist DHβE (50 μM). Superfusion of α-CtxMII did not block the inhibition caused by 0.1 μM nicotine in the NAc shell, however, superfusion of DHβE significantly inhibited the DA signal about 40% from baseline (Fig. 1D). Additionally, DHβE blockade of nicotine induced DA reductions was significantly increased when compared to α-CtxMII experiments. Using FSCV, we evaluated the effects of nicotine (0.01 – 10 μM) on evoked DA release in the NAc core. Nicotine significantly decreased the peak amplitude of the DA signal with an IC50 of approximately 0.1 μM (Fig. 2A,B; 0.01 μM, 0.1 μM, 1.0 μM, 10.0 μM). We noted varying results at higher concentrations of nicotine (10 μM) in the NAc shell (Fig. 1B) vs the NAc core (Fig.2B). The frequency dependency for nicotine effects on DA release in the NAc core (Fig. 2C) was similar to the shell; however, the maximum release occurred at 20 Hz rather than 40 Hz. 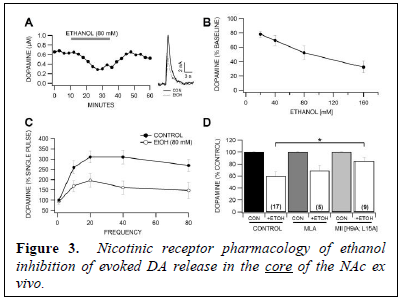 Nicotine significantly decreased the amplitude of the DA signal across all frequencies tested. We analyzed the effects of the α6 antagonist α-conotoxins α-CtxMII (100 nM) and α-CtxMII [H9A; L15A] on nicotine inhibition of evoked DA release in the NAc core. Superfusion of α-CtxMII significantly inhibited the DA signal about 30% from baseline while the more specific α6* antagonist α-CtxMII [H9A; L15A] only slightly inhibited the DA signal 8% from baseline. Both of the α-conotoxins blocked the inhibition caused by 0.1 μM nicotine in the NAc core (Fig. 2D).
Nicotine significantly decreased the amplitude of the DA signal across all frequencies tested. We analyzed the effects of the α6 antagonist α-conotoxins α-CtxMII (100 nM) and α-CtxMII [H9A; L15A] on nicotine inhibition of evoked DA release in the NAc core. Superfusion of α-CtxMII significantly inhibited the DA signal about 30% from baseline while the more specific α6* antagonist α-CtxMII [H9A; L15A] only slightly inhibited the DA signal 8% from baseline. Both of the α-conotoxins blocked the inhibition caused by 0.1 μM nicotine in the NAc core (Fig. 2D).
We then evaluated the effects of ethanol (20 – 160 mM) on DA release, as well as the effects of α-CtxMII (100 nM) on ethanol inhibition of DA release in the NAc core. Ethanol significantly decreased the peak amplitude of the DA signal with an IC50 of approximately 80 mM (Fig. 3A,B; 20 mM, 80 mM, 160 mM). Ethanol significantly decreased the amplitude of the DA signal across all frequencies tested with the exception of single pulse stimulation (Fig. 3C). This confirms the frequency dependent nature of ethanol in the NAc reported previously (Yorgason et al., 2013). We analyzed evoked DA release in the presence of ethanol with the specific α7 and α6 nAChR antagonists methylylcaconitine (MLA; 100 nM) and α-CtxMII [H9A; L15A] (500 nM), respectively. MLA did not alter ethanol inhibition of evoked DA release in the core at 80 mM ethanol (Fig. 3D). However, superfusion of the specific α6*-nAChR antagonist α-CtxMII revealed a significant effect of α-CtxMII [H9A; L15A] on ethanol inhibition of evoked DA release in the NAc (Fig. 3D).
Discussion
We demonstrated that evoked DA release is reduced by nicotine application in the NAc shell and core ex vivo. These inhibitory effects of nicotine were observed across all frequencies and were blocked by nAChR antagonists. Specifically, in the shell, nicotine’s effects were blocked by the α4β2* selective antagonist DHβE, but not by the α6 nAChR antagonist α-CtxMII. In contrast, nicotine modulation of DA signals in the core appears to be more through α6 nAChRs, as α-CtxMII, and the more selective α6 antagonist α-CtxMII [H9A; L15A], blocked nicotine’s inhibition of evoked DA release. We demonstrated that ethanol reduces stimulated DA release in the core in a frequency dependent manner, with greatest inhibition at higher stimulation frequencies. Ethanol’s inhibitory effects were blocked by α-CtxMII [H9A; L15A], but not MLA, suggesting that ethanol is acting through heteromeric a6* nAChRs, and not α7 homomeric receptors. Nicotine decreased evoked DA release ex vivo in both the shell and the core of the NAc with an IC50 of 0.1 μM. However, at the highest concentration of 10 μM, nicotine in the NAc core demonstrated significantly less inhibition of DA release (Fig. 1B, 2B), suggesting that the nAChRs in the NAc core desensitize to high concentrations of nicotine as compared to nAChRs in the NAc shell. The variability in response to the higher concentrations of nicotine in the NAc core vs NAc shell demonstrates specific differences in nAChRs subunits in the NAc core and the NAc shell. Our experiments with the specific α6*-nAChR antagonists α-conotoxins in both the NAc core and NAc shell demonstrate a distinct difference in the nAChR distribution in these anatomical regions. Given that α-CtxMII completely blocks the effects of nicotine in the NAc core and does not block the effects of nicotine in the NAc shell, yet DHβE completely blocks the effects of nicotine in the NAc shell, these results indicate that the NAc core primarily operates via α6*-nAChRs and the NAc shell primarily operates via α4β2 nAChRs.
Similar to nicotine, ethanol significantly decreased DA release and reduced phasic responses across all frequencies equally. Superfusion of the α7-subunit specific antagonist MLA had no effect on ethanol inhibition of evoked DA release, suggesting that ethanol is not acting in either the NAc core or NAc shell via α7*-nAChRs. However, as α-CtxMII and α-CtxMII [H9A; L15A] significantly reduced ethanol inhibition of DA release in the NAc core, suggesting that ethanol is acting via α6*-nAChRs in the NAc core. Thus, as α-CtxMII significantly blocked both nicotine and ethanol in the NAc core, both nicotine and ethanol appear to be acting via the same α6*-nAChRs.
Conclusion
These experiments verify that both nicotine and ethanol reduced phasic release of DA in the NAc, and that α6*-nAChRs are involved in both nicotine and ethanol inhibition of evoked DA release in the NAc core but not the NAc shell of C57BL/6 mice. This helps us to further understand the co-dependence of nicotine and ethanol and displays the mechanisms through which both addictive substances act, helping us to further investigate ways in which we could prevent this addiction and eventually, with further research, reverse its effects.