
Jacob Lewis and Dr. Thomas Knotts, Chemical Engineering
Main Text
Proteins are made up of extremely long chains of amino acids. Depending on it constituent amino acids, proteins fold in many 3-dimensional patterns to facilitate various biochemical processes such as catalyzing chemical reactions which allow us to live. How proteins fold is thought to be the “holy grail” of biochemistry research and, as of yet, no one has been able to predict exactly how a given sequence of amino acids will fold a priori. Protein folding has been probed in experiment and simulated on computers, but there are discrepancies between the two methods. Naturally, the experimental results are what computer simulations aim to achieve as a test of their validity. Two major discrepancies between computer simulations and experiments are the narrowness of the protein folding transitions and the number of transitions seen in larger proteins.
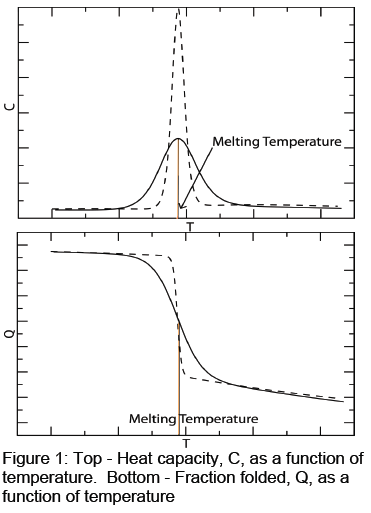
Protein folding transitions for thermally induced unfolding are marked by plotting the heat capacity of a protein as a function of temperature (Figure 1, top) or by plotting the fraction folded of the protein as a function of temperature (Figure 1, bottom). At low temperatures, the protein is considered fully folded, while at higher temperatures, the protein is considered completely unfolded. The center of the transition is considered to be the “melting point” of the protein (Note: this is not a phase transition). Experimental results often have sharp transitions, such as those seen in Figure 1 with dashed lines. Computer simulations more often produce the solid line transitions seen in Figure 1. The sharpness of the protein folding transition reflects its cooperativity. A high degree of cooperativity shows a sharp transition, like those seen in experiment. A low degree of cooperativity shows a broad transition like those seen in computer simulations.
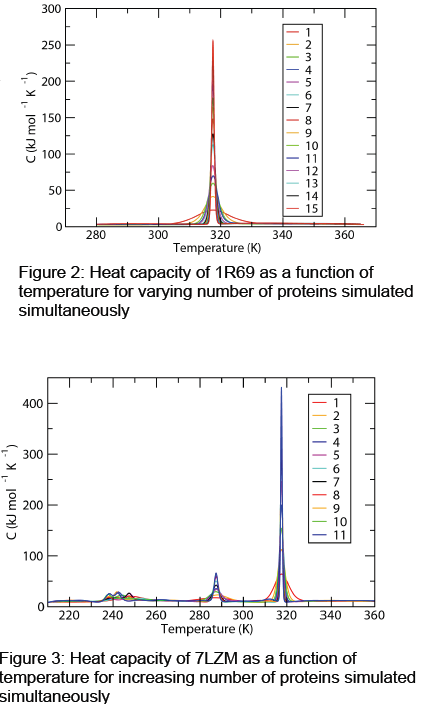
The computer simulations conducted in this research sought to bridge this discrepancy between computer simulated protein folding and experimental results by trying to simulate a system more like the experimental system. Typically, computer simulations are conducted on one single protein. Periodic boundary conditions are in effect, but a well-designed simulation does not allow interaction between a molecule/protein and its periodic images. In experiment, the systems measured contain many orders of magnitude more proteins. For example, a 1 μL sample of 1 mM concentration contains approximately 6 x 1014 proteins! Since current technology does not allow the simulation of that many proteins, we simulated up to 15 simultaneously of a small protein, 1R69, and up to 11 of a larger protein, 7LZM.
We found that as the number of proteins was increased for the smaller protein, 1R69, its single melting transition became increasingly narrow. As can be seen in Figure 2, the cooperativity of even 15 proteins is much greater than that of a single protein. The same trend was seen in the fraction folded curve as a function of temperature for 1R69.
For 7LZM, experimental results showed the protein having 1 or 2 transitions depending on the measurement used [1]. Simulating a single protein gave 3 distinct transitions, but as the number of proteins was increased to 11, one of the transitions did not become sharper and virtually disappeared. As seen in Figure 3, the lowertemperature transition became jumbled instead of sharper, while the middle transition became slightly sharper and the higher-temperature transition became much sharper. Closer inspection of the intermediate states between transitions for this protein show that the lowertemperature transition is uncooperative on the molecular level, since it essentially represents a small part of the protein becoming unhinged. The higher temperature transition represents the major unfolding event and is therefore the most cooperative. Simulating a larger number of proteins arguably brings the simulation of the folding of 7LZM into agreement with experimental results, something that is untrue of simulations with the single protein.
The results of this experiment were more detailed than can be described in this report, but they represent an important step in trying to accurately simulate real-world biological processes. It is unlikely that the number of proteins being simulated simultaneously is the only reason for the discrepancies between experiment and simulation, but it does represent a major discrepancy that will contribute to the end goal of a priori prediction of protein folding. This experiment and results were presented on November 9th, 2010 at the American Institute of Chemical Engineers Annual Conference in Salt Lake City, UT. Another presentation of this research is scheduled for the 2011 Utah Conference on Undergraduate Research at Weber State University in February. A paper detailing this research is in preparation to be submitted for publication to Physical Chemical Letters in January.