
Matthew Whited and Dr. Joshua L. Andersen, Department of Chemistry and Biochemistry
In cancer patients, a lack of cell apoptosis (programmed cell death) coupled with increased levels of cell proliferation leads to the formation of a tumor. Among the available treatment options, cytotoxic chemotherapy is the most common. The primary purpose of this treatment is to induce tumor cell apoptosis. In many cases, tumor cells develop chemoresistance, rendering chemotherapy ineffective as a method to activate apoptosis. Understanding the mechanisms that lead to chemoresistance is a critical step toward developing methods to increase the effectiveness of chemotherapy treatments.
Within most solid tumors are regions of intermittent hypoxia (cycles of hypoxia and reoxygenation). Since tumor cells are not inherently programmed for such an environment, they must either adapt to low oxygen or die. Tumor cells that adapt to hypoxia are often markedly chemoresistant, while those that die promote tumor shrinkage. The death of tumor cells in response to intermittent hypoxia is likely caused by the generation of cell- damaging reactive oxygen species (ROS) that rapidly spike during periods of reoxygenation.1 Thus, we posit that the ability of tumor cells to endure high levels of ROS determines whether they live or die in response to intermittent hypoxia.
Superoxide dismutase-1 (SOD1) is an enzyme that protects cells from ROS generated by hypoxia/reoxygenation.2 It does this by converting toxic superoxide ions into hydrogen peroxide and oxygen. SOD1 is expressed at high levels in various tumors and has recently been shown to be a promising therapeutic target in lung cancer where its expression promotes tumor survival.3 However, the mechanisms by which SOD1 activity is regulated in cancer are not understood. Previous data from Dr. Andersen’s laboratory suggests that acetylation of SOD1 may modulate SOD1 function.4 Moreover, in a proteome-wide study to understand the role of protein acetylation in the tumor cell response to hypoxia, Dr. Andersen found that acetylation of a specific lysine residue (lysine 123) in SOD1 was high in cells that eventually succumb to hypoxia and die. Conversely, cells that survive hypoxia have undetectable levels of SOD1 acetylation.
These data suggest that the acetylation status of SOD1 may influence whether cells survive intermittent hypoxia. Specifically, we hypothesize that acetylation of SOD1 at lysine 123 reduces its ability to lower ROS levels, thereby rendering SOD1 less able to promote tumor cell survival through periods of intermittent hypoxia. To test this hypothesis, we performed site-directed mutagenesis, using a QuikChange Mutagenesis kit, in order to introduce K123Q and K123R mutations into the SOD1 protein. Mutation of lysine 123 to arginine mimicked a deacetylated lysine, while the mutation to glutamine mimicked an acetylated lysine. After obtaining both mutations, we transfected 293 cancer cells with the K123Q mutant form of SOD1. We then used a Superoxide Dismutase Assay Kit from Cayman Chemical Company to determine the activity of the SOD1 protein with the Q mutation, as well as wild-type SOD1. Additionally, we expressed wild- type SOD1 under both hypoxic (lacking oxygen and glucose) and normoxic (normal oxygen and glucose) conditions to determine if SOD1 acetylation was altered. Using Western Blot analysis and treating with an acetyl antibody and accounting for loading, we determined the relative amount of acetylation between hypoxic and normal conditions.
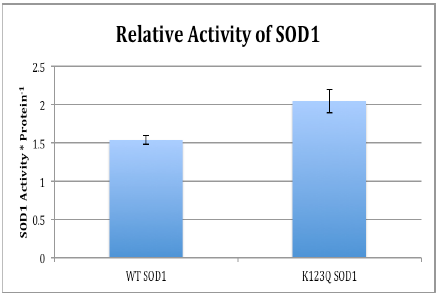
Preliminary results from the SOD1 Activity assay revealed that the activity of SOD1 was increased in the 293 cells that were expressing K123Q SOD1. Remember that this mimics the acetylated version of SOD1. Data was obtained through an activity assay specifically designed to correlate SOD1 activity with absorbance. From the absorbance values obtained, the activity was ascertained and adjusted for loading.
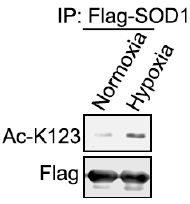
Results from the Western Blot clearly indicated that under hypoxic conditions, acetylation of wild-type SOD1 was increased by the 293 cancer cells. This increase in acetylation reveals how the cells are responding to hypoxic conditions, which as discussed earlier, mimics the internal parts of the tumor that have inadequate blood flow and access to resources, such as glucose.
One of the ways that hypoxic conditions affect cells is by increasing the abundance of oxidative stress. As such, a tumor cell’s ability to increase acetylation should, as indicated by the preliminary results from the SOD1 activity assy, should allow it to remove harmful free radicals and allow the tumor cell a greater chance of survival. In conclusion, more repeats of the SOD1 activity assay need to be conducted to determine if the difference between wt SOD1 and K123Q SOD1 is reproducible. Additional experiments with the K123R SOD1 mutant will further add to our understanding of how the deacetylated version of SOD1 functions in the cell. Due to these exciting findings, we will continue to pursue SOD1 in order to learn more about the effects of acetylation as a post-translational process.
References
- Brown NS, Bicknell R. (2001) Hypoxia and oxidative stress in breast cancer. Oxidative Stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 3(5):323-‐7.
- Morita-‐Fujimura Y, Fujimura M, Yoshimoto T, Chan PH. (2001) Superoxide during reperfusion contributes to caspase-‐8 expression and apoptosis after transient focal stroke. Stroke. 32(10):2356-‐61.
- Somwar R, Erdjument-‐Bromage H, Larsson E, Shum D, Lockwood WW, Yang G, Sander C, Ouerfelli O, Tempst PJ, Djaballah H, Varmus HE. 2011. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc Natl Acad Sci U S A. 108(39):16375-‐80. Epub.
- Andersen JL, Thompson JW, Lindblom KR, Johnson ES, Yang CS, Lilley LR, Freel CD, Moseley MA, Kornbluth S. (2011) A biotin switch-‐based proteomics approach identifies 14-‐3-‐3ζ as a target of Sirt1 in the metabolic regulation of caspase-‐2. Mol Cell. 43(5):834-‐42.