Lars P.C. Nielsen and Dr. Morris J. Robins, Chemistry and Biochemistry
Glucose transporter proteins constitute one class of transmembrane integral proteins that have received much attention in the current chemical literature. Many prominent groups have set out to explore their mechanisms, kinetics, and active sites. Studies have focussed on one of these in particular, Human Glucose Transporter I, the most common and efficient at mediating cellular glucose concentrations. Studies have also proven that certain nucleoside transporters exist, analogous to the sugar proteins, which regulate nucleoside and nucleotide concentrations in cells.
Just as with Human Glucose Transporter I, one nucleoside transporter, known as the human intestinal es protein, dominates the regulation of intracellular nucleoside concentrations. It is the most efficient and extensive, and therefore, the most important in the human body. Because of recent progress in cloning human es transporter, we anticipate intense research on these proteins in the years to come.
The rate of cellular replication in some types of cancer cells is higher than in related noncancerous cells. Because nucleosides and nucleotides play fundamental roles in replication as genetic building blocks, the most common clinical approach for treating various cancers involves administration of drugs (often nucleoside analogs) designed to inhibit or slow duplication of genetic material. Cells in a state of replication will uptake such nucleoside analogs more readily than cells not actively involved in cell division. Unfortunately, this approach to cancer has side effects; these drugs kill quickly dividing cancer cells, but can also be toxic to normal cells.
Many cells in the body enhance their nucleotide pools through facilitated diffusion mediated by specific nucleoside transport proteins. Although there is an array of such proteins, the human es transporter is responsible for most nucleoside uptake. Unadkat and coworkers have characterized the human intestinal es nucleoside transporter and have suggested possible profiles for its inhibition. However, they note that “none of the studies using human cells included a comprehensive array of nucleoside analogs where systematic substitutions on the sugar or the base or both were included.” A complete correlation between binding pocket affinity and key structural features of nucleoside derivatives has not been possible before due to difficulty in cloning the transporter and lack of available radioactive substrates. According to Nature Medicine, “the molecular cloning of the first human nucleoside transporter represents a milestone in the study of both nucleoside pharmacology and chemotherapeutics.”
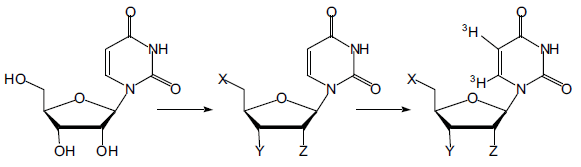
My research project involved the synthesis of various uridine nucleosides systematically substituted at different positions of the sugar moiety. These compounds have be sent to collaborators at the University of Alberta who will label the pyrimidine base with tritium. The labeled compounds will then be analyzed with the cloned es protein along with three additional transporters cloned and expressed at the University of Alberta under the direction of Professor C. E. Cass, a world leader in nucleoside transport and molecular biology.
We have chosen an array of twenty-two compounds logically varied at the 2´, 3´, and 5´ positions of the ribosyl moiety that should provide enough information to effectively deduce the structural features necessary for tight binding. Seven of these potential inhibitors are commercially available. I have made and sent an additional five compounds over the past five months. Many others are still being made and will be synthesized over the next eight months. Our research will set the precedent for the synthesis and analysis of such compounds with the transporter proteins.
Our research may have far-reaching clinical implications. For example, some strains of leukemia depend on different modes of nucleoside transport (i.e., modification of the sodium pump). A strong blocker for the normal human es transporter could theoretically prevent the harmful ingestion of nucleoside drugs by normal human cells without preventing uptake by leukemia cells. This would reduce adverse side effects even at drug dosages commonly used in chemotherapy. Also, the inhibition of transport of natural uridine and deoxyuridine substrates will play an important role in the design of potent inhibitors against other transporters.
The mechanism of the es transporter is not well characterized, and therefore, the compounds that I am still making may help profile nucleoside transporter activity and inhibition. It is expected that the characterization of the protein’s binding site will provide a data base for the design of cancer treatments. We anticipate vigorous collaborative studies that will result in publication of several papers in reputable organic and biochemistry journals.
Very quickly I would like to outline the synthesis of a few compounds I made. 5′-chloro-5′- deoxy-uridine was made by protecting 2′,3′ with a sulfinyl ester followed by chlorination of the remaining hydroxyl group. This compound was radically reduced using tributyltin hydride and azobisisobutyronitrile. 5′-azido-5′-deoxy-uridine was made by nucleophilic displacement of the chloride with sodium azide in dimethylformamide. 2′,5′-bis-O-TBDMS-uridine was made by treating uridine with tert-butyldimethylsilylchloride in pyridine. This compound was separated from its 2′,3′ diastereomer by silica column chromatography. Treatment of the former with phenoxythiocarbonyl chloride gave the tri-O-protected nucleoside which was also radically reduced and desilylated with ammonium fluoride to give 3′-deoxyuridine. Diphenyl carbonate was also added to dry uridine in hexamethylphosophoramide with a catalytic amount of sodium bicarbonate. This formed a cyclic leaving group which was displaced by the pyrimidine to form the 2′-cyclonucleoside. This cyclonucleoside was treated with sodium hydroxide to hydrolyze the base and form the arabinofuranosideuracil. Azide displacement of the cyclonucleoside was also possible and gave the 2′-azido-2′-deoxyuridine. This outlines the synthesis of a few compounds that constitute only a part of this continuing project.