
Jeffrey Stephens and Dr. Merritt Andrus, Chemistry and Biochemistry
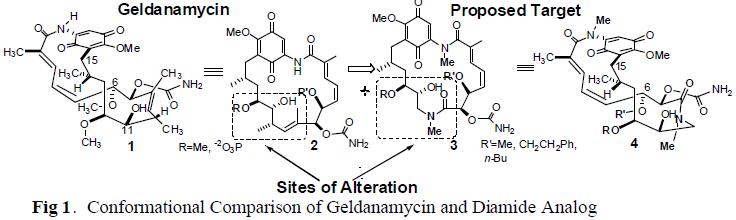
Cancer is a condition unlike any other known to man. It is one of the leading causes of death in the United States. In 2000, over 500,000 deaths resulted from various cancers.1 This has led to an extensive search for improved ways to treat cancer, and has left a daunting challenge to develop new ways to treat this condition. The natural product Geldanamycin (GA), the most potent of the benzoquinone absamycin antibiotics, has shown to have great promise as an anti-tumor agent. The analog of Geldanamycin, 17-Allylanino-GA, is currently in clinical trials2, and the first total synthesis of GA was recently completed in the Andrus Labratories.3 GA binds heat shock protein 90 (Hsp90), a 90 kDA protein essential for the development of new proteins in developing cells, whereas mature cells remain unaffected. A recent publication in Nature4 describes bioassays performed using 17-Allylamino-GA, wherein the drug has been shown to bind Hsp90 up to 100 times more tightly in cancer cell lines than in normal cell in vivo. A new analog is currently under development in the Andrus Laboratories, which could potentially increase the binding to Hsp90 with even greater affinity than ever, and decrease the toxicity of Geldanamycin to human biological systems.
Initial production of the target drug has been met with complications. Starting with the methoxyhydroquinone 5, the alcohol groups on the benzene ring were protected with SEM with a yield of 90 %. Formylation using DMF and BuLi allowed for the formation of 7 by placing the aldehydes group on the ring in the proper position, giving a yield of 61%. However, the next step led to complications.
The nitration of the benzene ring has not gone as hoped. It seems that in the nitration step, the desired product is recovered in a low yield. Additionally, one of the SEM groups on the alcohol seems be falling off under the strong acidic conditions of the reaction, which forms compound 8. Milder conditions have been attempted, but nothing has proved to be very successful.
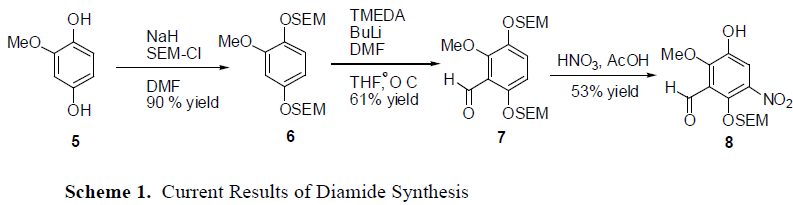
The use of acetic acid with nitric acid is similar to the conditions employed in the original synthesis of Geldanamycin. Various other conditions have been explored, but with poor results. Scandium triflate with acetic anhydride was also attempted, but with an even lower yield than before. A nitrnium tetrafloro borate with a picoline was also attempted, but also had diasterous results. None of these reagents allowed for the SEM groups to stay on.
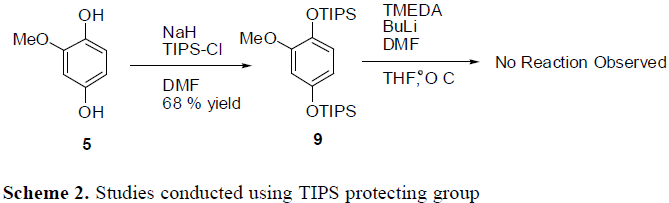
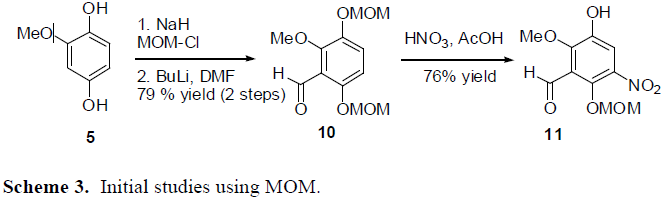
To get around this problem, other protecting groups were explored. The use of TIPS on the oxygen groups of the benzene ring was attempted. The TIPS groups went on fine, but attempting to put the aldehydes on the ring at this point led to extremely low yields. It seems that the TIPS groups are just too large to effectively allow for any further reactions on the benzene ring. Another solution that is yet to be attempted is to use the MOM protecting group. Initial studies have been conducted using MOM. The formation of the aldehydes and of the nitro group seems to proceed effectively. However, one of the MOM groups do fall off during the nitration step, but re-protection occurs in high yield. It seems that the MOM protecting group may be used for the synthesis of the target drug.
Additionally, studies were conducted and completed in the development of a new methodology that will be used later in the production of the diamide drug. A phase-transfercatalysis reaction has been developed using a glycolate substrate that will allow fo r alkylations to be performed quickly and allows for high selectivity of the desired stereocenter. Such reactions can be performed quickly and effectively. A wide variety of electrophiles may be employed, allowing for the possibility of numerous new attachments that can be added to a carbon skeleton. After the alkylation, the diphenyl protecting group on the alcohol can be cleaved, and the ketone oxidized to an ester and also cleaved to form the carboxylic acid. Synthetically, this allows for greater flexibility in any synthesis in that a stereocenter can be fixed into its proper position and any carbon group can be added. This is a key step in the synthesis of the diamide analog, and will reduce the number of steps required to create the compound.
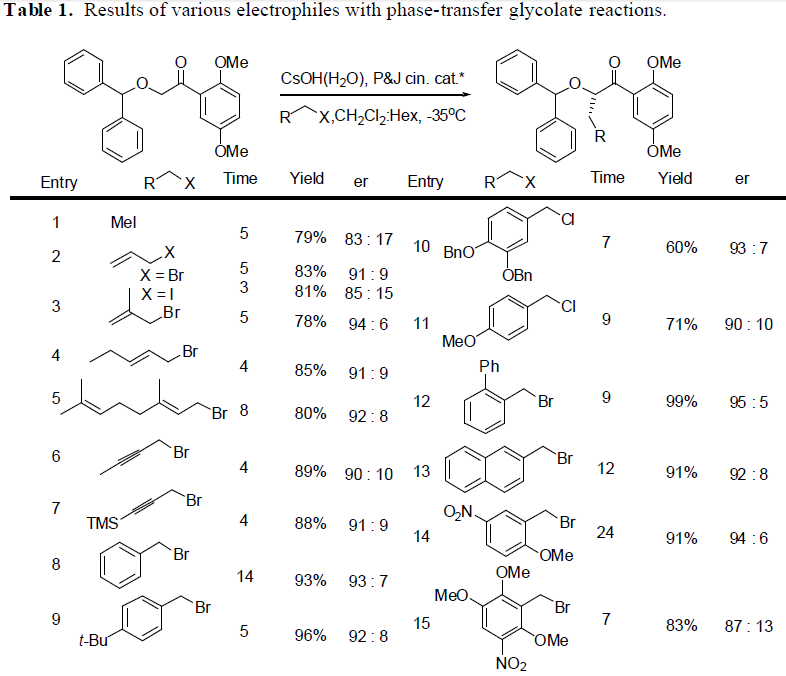
The table shows the test compounds that maybe employed in an alkylation using phase transfer conditions. In synthesizing the diamide target, the same substrate will be used, but an aldol reaction will be conduced instead of a alkylation. This will place both of the alcohol stereo centers in place in the same step. This is the progress of the research of this proposal to this point.
