
Joshua Wilkerson and Dr. Thomas Knotts, Chemical Engineering
Introduction
Proteins are versatile biological molecules that catalyze chemical reactions, provide cellular structure, and transport materials throughout the body. Proteins have been adapted for use in a wide variety of industries including food processing, medical diagnostics, and therapeutics. However, proteins must often be modified to enhance their utility in these operations. One method that can be used as an initial step in protein modification strategies is replacement of a naturally occurring amino acid with an unnatural amino acid (uAA). Previous research has suggested that it is very difficult to predict the stability of a uAA-modified protein based on any individual factor. However, a computational model has been used to overcome this limitation by testing each site by creating a coarse-grain model of the modified protein and simulating it. My project furthered this idea. The goals of my project were: 1) experimentally determine which AA of a protein can be replaced by uAAs without compromising protein stability and activity and 2) use a new computational molecular model to simulate proteins with uAAs and predict behavior.
Methodology
My project utilized a combined experimental/computational approach to gain deeper insights into the underlying factors affecting uAA-substituted protein stability. T4 lysozyme was the protein I tested throughout my project. Because it has been studied both computationally and experimentally and is well understood compared to many other proteins, it presented a good starting point for my research. Various sites were chosen for modification by uAA. Sites were chosen at locations throughout the protein in regions of varying structure. The types of structure at the locations chosen included alpha helices, beta sheets, linker regions, and unstructured regions. The proteins with the uAA modification were created by QuikChange mutagenesis and cell free protein synthesis.
Protein production requires a DNA template. DNA primers are used as part of the DNA replication process to produce enough DNA template for protein synthesis. QuikChange mutagenesis is a process of DNA modification that can be used to produce a modified DNA template. It involves using primers that have a slight mismatch in order to modify the sequence. I created primers with a mismatch for each of the sites chosen and produced DNA template to make a protein modified with a uAA at each of these sites. Cell-free protein synthesis (CFPS) is a method of producing proteins by lysing cells to obtain their protein-making machinery. It was optimal for this project because it allows direct access to the reaction environment. This was very useful, because incorporating uAAs into a protein requires some components not found in a typical cell. They were easily added because CFPS was used. This portion of the experiment is ongoing. Some of the sites have been synthesized through CFPS already, while others will be completed in the upcoming months.
After CFPS, the protein created was test by the Protein Thermal Shift Assay. The Thermal Shift Assay uses a fluorescent dye that binds to certain residues. The protein is heated, and as the temperature rises, more of these residues are exposed, allowing binding of the dye. Data from this assay can be analyzed to determine the temperature at which the protein becomes unstable. All sites that have been produced by CFPS have been tested with this assay, and the others will be tested as they are produced. Computer simulation has been delayed because of difficulty in creating parameters to properly represent the uAA modification. A graduate student in Dr. Knotts’ lab is currently doing this work as part of his PhD. Parameters for all-atom simulation are currently being validated, and we will then convert them to coarse-grain parameters. This will take place in 2019. I am trained and ready to perform simulations once we have the parameters.
Results
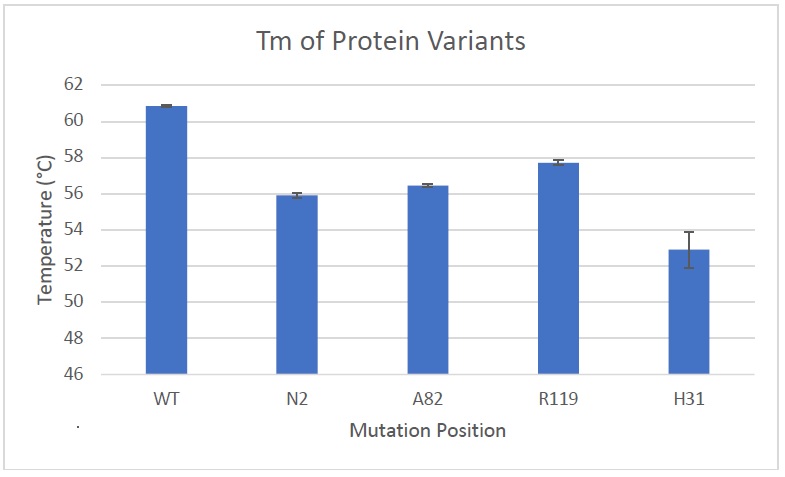
The attached graph shows the stability of the protein mutated at 4 positions, compared with the wild type protein (no mutation, labelled with “WT”).
Discussion
The attached results show that all sites lead to destabilization of the protein, but to varying degrees. In particular, H31 is more destabilized than the others. This variety in stability continues to suggest the importance of a computational screen to find the best locations for protein modification.
Conclusion
The results obtained further confirm our previous findings: a single parameter is not sufficient to predict uAA-incorporated protein stability. When it is ready, we expect our computational model to provide data that closely mirrors that obtained experimentally. This work has helped improve a tool to site-specifically select sites for uAA incorporation, paving the way for better protein therapeutics