
Allen Weinert and Scott Weber, Microbiology & Molecular Biology
Introduction
The leading cause of dementia in elderly patients is Alzheimer’s disease (AD), a degenerating and fatal neurodegenerative condition. AD is a proteopathic disease caused by extensive accumulation of amyloid beta plaques and neurofibrillary tangles. A recent genomewide association study analyzing 59 AD-associated cerebrospinal fluid (CSF) samples statistically associated chemokine receptor mutant CCBP2-V41A with increased CSF protein levels of the proinflammatory chemokine CCL2. CCBP2 is a known binding partner of CCL2. We hypothesized that CCBP2-V41A receptor alters CSF levels of CCL2 and that raised CCL2 levels alters immune cell function, resulting in amyloid beta deposition in the brain (Figure 1).
Methods
To test our hypothesis that CCBP2-V41A will bind to CCL2 less than the wild type, we determined to compare the receptor expression of cells that we have tested with the antibody, to cells that were first incubated for an hour with CCL2 before testing with the antibody. If our hypothesis is correct, we should see a larger reduction of expression in the wild type than the mutant. This signals that there was better binding in the wild type and thus more blocking of the receptor. To test why there could possibly be a difference in binding, we have run chimera software to analyze the protein shape and what differences there could be between the wild type and mutant. We have also run metabolism analysis to test if there is a larger oxygen consumption rate in the wild type than the mutant when ATP synthase is blocked and then uncoupled under activated conditions.
Results
In the competitive binding assay, we saw a 27.2% drop in receptor expression in the wild type when incubated with CCL2 whereas the mutant only had a 10.4% drop (Figure 2). This finding suggests that the wild type binds better with CCL2 than the mutant and is consistent with our hypothesis. The chimera analysis did not reveal any changes in the receptor protein structure or in hydrophobic interactions (Figure 3). The oxygen consumption rate analysis of the cells in the wild type and mutant cells showed no difference in ATP production under activated conditions (Figure 4). We will measure glycolysis next since recent research has suggested that chemokine activation affects glycolytic metabolism more than mitochondrial respiration.
Discussion and Conclusion
Our preliminary data suggests that there is a difference in binding between the mutant and wild type chemokine receptors. We are still unsure how the single amino acid change causes this, but the data does support our hypothesis that the CCBP2 mutant does not bind as well to CCL2 and thus cannot scavenge CCL2 out of CSF as well which would cause an increase of inflammation. Recent research has shown that inflammation is most likely the main cause of Alzheimer’s disease. With this information and further testing, it is possible that this mutation could be a genetic marker of Alzheimer’s disease and enable the development of novel immunotherapies. From here we hope to test glycolytic metabolism as well as repeat the competitive binding analysis before publishing.
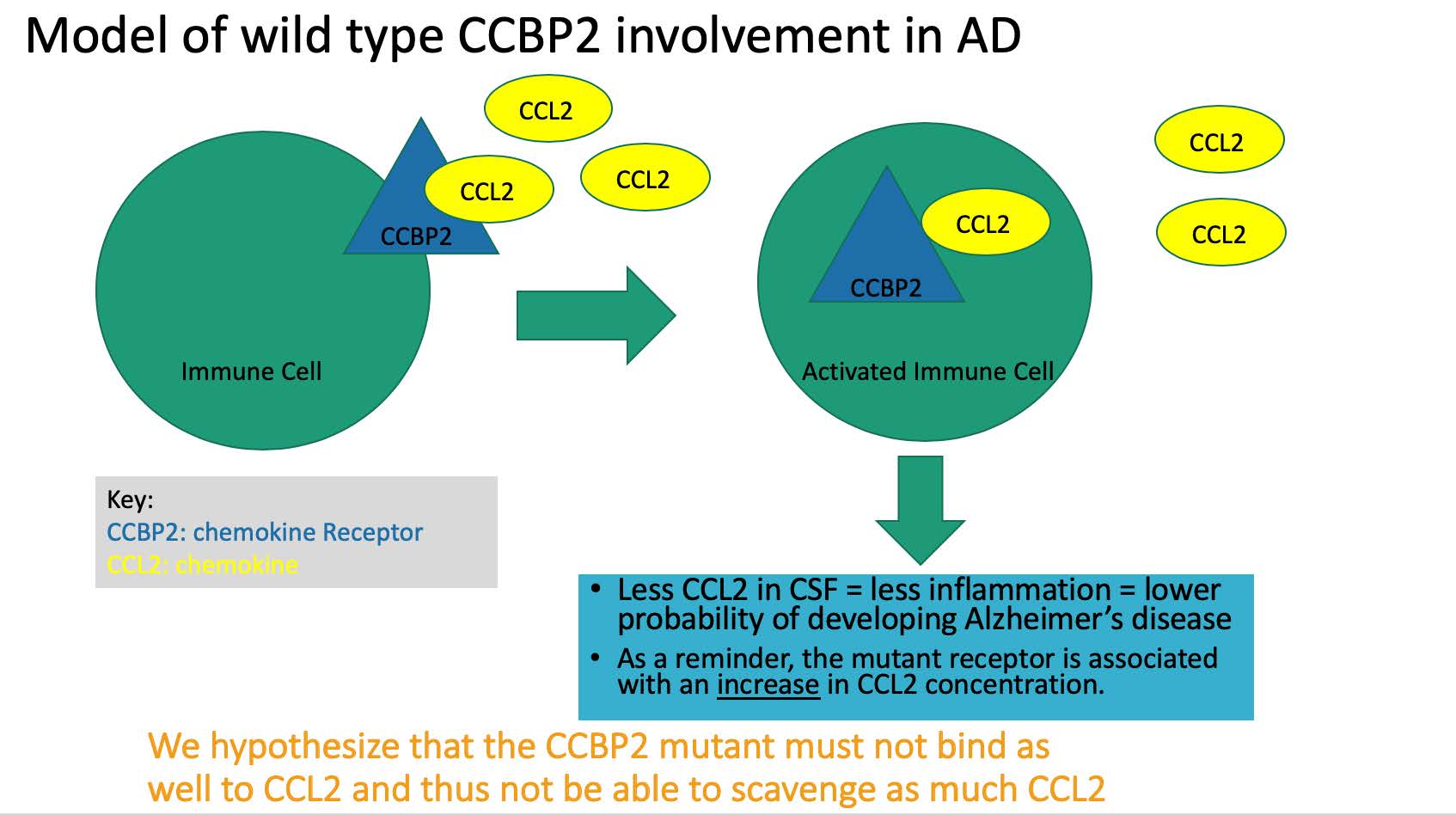
Figure 1: Model of what the normal wild type chemokine receptor’s role in AD is and highlights the reason the mutant could be making disease progression worse.
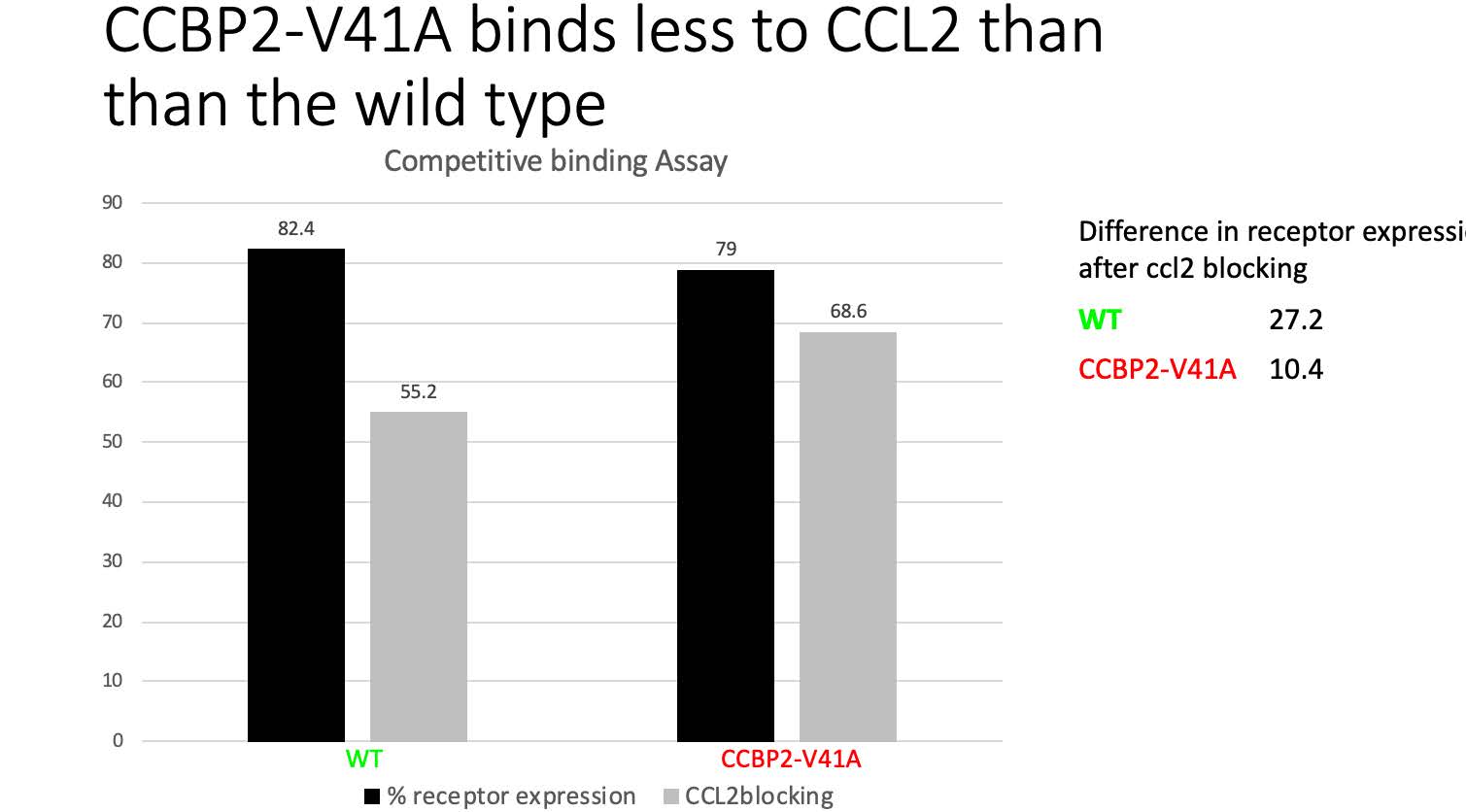
Figure 2: Binding assay showing that the mutant (red) has 16.8% less binding to CCL2 than the wild type (green) receptor.
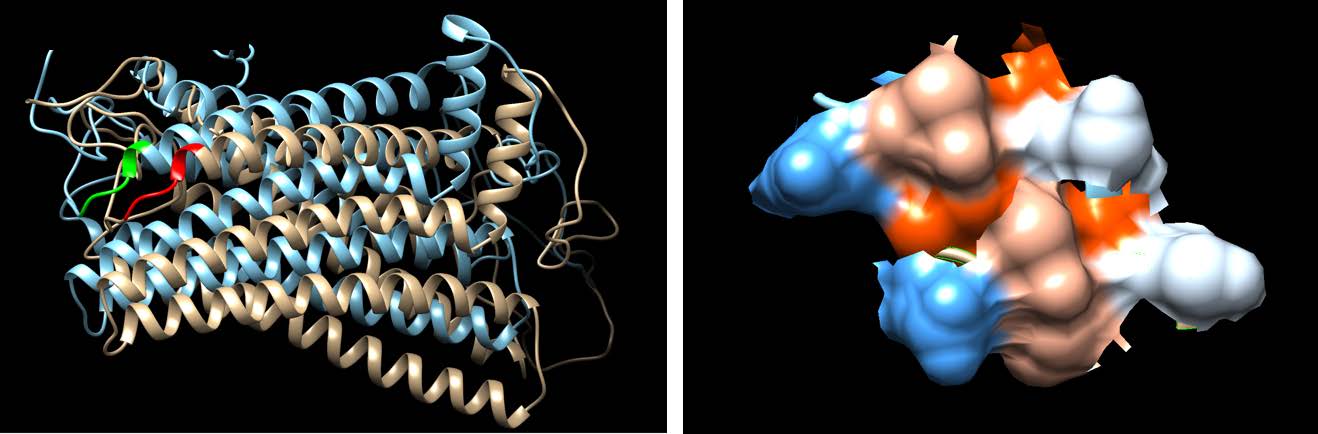
Figure 3: Images created via protein structure software to analyze differences between wild type (blue) and mutant (tan). No notable differences found.
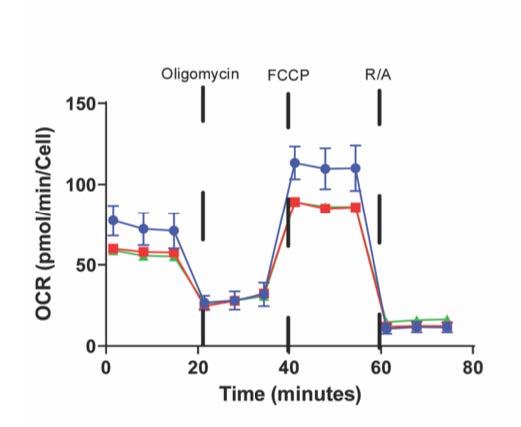
Figure 4: A metabolism assay that shows no difference in energy synthesis between wild type (green) and mutant (red). The control is shown in blue.