
Aaron Kubosumi
Microwave Promoted Iminyl Radical Cyclization
Dr. Steven L. Castle
Department of Chemistry and Biochemistry
Introduction
Pyrrolidines and their derivatives are found in many biologically active
compounds, such as Lipitor and Atorvastatin, which possess a wide variety of remarkable
pharmaceutical properties. Therefore, the efficient synthesis of functionalized pyrrolidines is of
great value to organic chemists. Multiple attempts have been made to synthesize compounds
containing pyrrolidine ring systems. Walton 1 utilized radical cyclization, via microwave heating,
to transform Ophenyl oximes with terminal alkenes to dihydropyrroles, compounds closely
related to pyrrolidines. Similarly, Sammis 2 proposed a method involving radical cyclizations of
azidederived aminyl radicals onto silyl enol ethers. Unfortunately, the method proposed by
Sammis requires highly toxic and explosive reagents. The synthetic route proposed by Walton
could in principle allow for an effective synthesis of important pyrrolidine ring systems, but to
date no one has explored this application. Furthermore, Walton’s synthesis involves H atom
trapping, which prevents access to highly functionalized compounds that could be obtained by
trapping with other atoms.
Our research group took Walton’s findings further by using TEMPO as the radical trap.
We were able to produce various functionalized pyrroles in good yields using different alkyne
precursors3 We have now turned our attention to what kind of stereochemistry the microwave
promoted iminyl radical cyclization process imparts. Most important biologically active
pyrrolidine derivatives, apart from possessing a wide range of functional groups, also have
specific threedimensional orientations. The ability to construct these compounds
stereoselectively would be important in future synthesis. Initial work performed by Yu Cai and
Zach Gibson showed that our process imparts unexpected orientations at stereocenters. My
current work builds upon this by 1) verifying the structure that Zach Gibson reported, and 2)
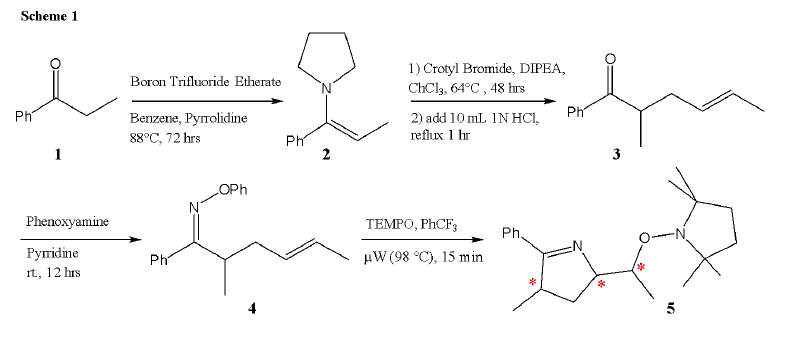
determining the stereochemistry of compound 5 (see Scheme 1 ). The work presented here will
cover the latter synthesis.
Methodology
Reactions were designed according to similar reactions found in scientific literature, among which includes our previously published work regarding microwave promoted iminyl radical cyclizations.
Results and Discussions
The synthesis of the compounds in Scheme 1 were completed and determined via MS and 1H NMR. Unfortunately, the Ophenyl oxime formation step provided trouble throughout the semester. The pure phenoxy amine, used in previous cyclization reactions, is a light colored oil which is then reacted with HCl in order to store it as the more stable, white amine salt. However, the addition of HCl, while initially producing the white salt, would over time (within the span of an hour) turn the salt a dark red color suggesting degradation of the product. Indeed, this red, impure salt was tested and would not form the oxime upon reaction with a ketone. For a time we produced phenoxy amine and stored it as the oil, albeit the red color was still present. Another student, Mary Jackman, spent time determining what impurity was eluting with the phenoxy amine and hypothesized that it was leftover Cu(I) from the first step chelating with the nitrogen. On a column, I was able to separate out a red oil from the phenoxy amine oil but 1H NMR analysis was unhelpful in determining the identity of this contaminant.
We discovered, however, that washing the phenoxy amine salt over filter paper with DCM and
filtering with vacuum washed out whatever impurity remained, leaving behind the pure salt. This
method was even able to rescue old phenoxy amine that was stored as oil in the refrigerator. By
reacting this salt with a ketone (propiophenone), we were able to determine that the salt indeed
functioned in forming the oxime product.
The 1H NMR spectrum of compound 5 showed more hydrogens than would be expected
in the pure product, indicating the presence of impurities. These impurities make us unable to
perform NOESY NMR experiments at the current time. However, analysis of the spectrum does
offer some insights. The apparent splitting of the hydrogen peaks ~4.2 in a 1:1 ratio suggest a 1:1
ratio of diastereomers at one of the stereocenters (which we hypothesize to be the stereocenter
immediately adjacent to the oxygen). We are currently in the process of assessing the purity of
our reagents and solvents. We are also testing different solvent systems, such as DCM in hexane,
to determine whether or not we can better purify each compound so that we do not have to use
the crude products as starting material in many of the reactions. The rationale behind using the
crude product, especially in the cyclization step, is that our past experience shows that the oxime
precursor and the cyclized product have vastly different rf values and therefore any impurity that
elutes with the oxime should not elute with the cyclized product. Indeed, that is the case with
compound 4 and compound 5 ( rf values of 0.9 and 0.5, respectively). However, we think thatperhaps whatever impurity is present may be fragmenting in the microwave and reacting with
other radical species present such as TEMPO. This hypothesis is supported by the presence of
multiple bands (around 7) on the crude TLC when only 3 are expected (TEMPO, oxime, and
product).
Conclusion
While we have successfully synthesized compound 5 , as shown by MS and 1H NMR, we have not yet been able to determine stereochemistry of compound 5 . Due to the
presence of around 12 unaccounted for hydrogens in the 1H NMR of the final product, we have
not been able to perform NOESY NMR experiments. We are currently searching for the cause of
these impurities so that we can remove them and get a clean 1H NMR spectrum and ultimately
perform a NOESY analysis so as to determine the three dimensional arrangement at the three
stereocenters in compound 5 .