
William Bradley
Faculty Mentor: Bradley Bundy, Dept. of Chemical Engineering
Introduction
As the search continues for novel proteins with market-incentivized properties, the demand for tools to accelerate the identification of these proteins increases. This has led to investigating methods for screening proteins that yield faster results (high-throughput) with greater titers of protein stable enough for analysis (high yield).
Cell-free protein synthesis (CFPS) offers an ideal testing ground for protein screening by preserving the cell machinery required to transcribe DNA into protein in an extract not bound by a cell wall. Well documented is the ability of linear DNA, or linear expression templates (LETs), instead of circular DNA, or plasmid, to serve as a template for protein production in CFPS. Yet while the use of LETs rather than plasmids reduces the time required to analyze DNA sequences (i.e. more high-throughput), past studies of proteins with LETs have observed lower yields of stable proteins, making analysis of some protein variants difficult and in some cases impractical. This lower yield is often attributed to the relative instability of LETs, which are thought to degrade quickly from exonucleases in CFPS, yet other differences in LET vs plasmid preparation may cause the discrepancy in protein expression levels.
The focus of this work was to adapt high-throughput methods of screening proteins to enhance the stability of LETs and thus yield of stable protein from CFPS. Specifically, various DNA purification methods were used and a base-pair ‘extension’ was added to protect LETs and thereby attain plasmid-level yields without losing the high-throughput benefits of LETs.
Methodology
For each experiment in this study, a DNA sequence encoding sfGFP was amplified with the polymerase chain reaction (PCR) using Taq polymerase. The primers with and without a 31 base pair extension were used in the PCR amplification. Replicated DNA was purified and concentrated via one of three methods: a commercial kit known to use guanidine hydrochloride (Gu-HCl) in its buffers (here Qiagen’s Miniprep purification kit was used), gel filtration via Sephadex G50 resin bed, and direct addition of DNA to cell-free extract without purification.
Escherichia coli extract was prepared using a BL21 (DE3) StarTM strain and the ‘energy source’ for CFPS via the PANOx system. All CFPS reactions were carried out according to a standardized protocol. Briefly, reagents were combined in a microcentrifuge tube for a final reaction mixture of 25% extract (v/v), 25% energy source (v/v), 12nM DNA, and magnesium glutamate with concentration optimized for extract (for this study: 10mM). These mixtures were aliquoted in 20uM droplets onto a 96-well opaque flat-bottom plate, sealed from air, and incubated for 8h at 37°C. Completed reactions were diluted with 40 uL water to cover bottom of wells before reading plates with Syngery Mx microplate reader. Protein yield was determined using a linearly correlated calibration curve.
Results
Figure 1 offers side-by-side comparisons of protein yields for different DNA sources and purification methods. Increased concentration of Gu-HCl decreases protein expression activity of CFPS with plasmid DNA source (1, 2, 3). LET purified with Gu-HCl containing buffers exhibited lower yields (4). Protein expression using LET was markedly improved when purified via gel filtration without Gu-HCl (5). Protein expression using LET was further improved to plasmid-based expression levels by introducing ~30 bases-long ‘protective’ sequences on either side during PCR amplification (6).
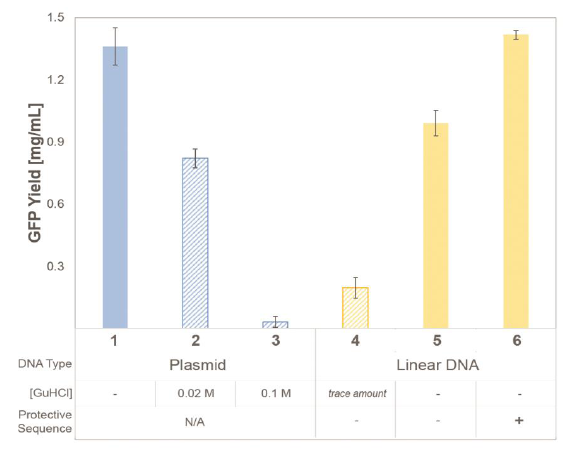
Figure 1 – Protein yields measured by fluorescent expression of active protein after CFPS reaction.
Figure 1 offers side-by-side comparisons of protein yields for different DNA sources and purification methods. Increased concentration of Gu-HCl decreases protein expression activity of CFPS with plasmid DNA source (1, 2, 3). LET purified with Gu-HCl containing buffers exhibited lower yields (4). Protein expression using LET was markedly improved when purified via gel filtration without Gu-HCl (5). Protein expression using LET was further improved to plasmid-based expression levels by introducing ~30 bases-long ‘protective’ sequences on either side during PCR amplification (6).
Discussion
We suspected that guanidine hydrochloride was a significant contributor to losses in protein yield due to its common use in DNA purification procedures based on high-throughput (requires <60min) silica adsorption. Gu-HCl is a chaotropic salt whose function includes denaturing protein. Our results indicate that Gu-HCl in sufficiently high concentrations will inhibit protein translation. Gel filtration such as Sephadex columns while equally high-throughput (requires
Also of interest was guarding LETs whose exposed ends render them vulnerable to exonuclease degradation. An extension sequence of 31 base pairs was added as buffer between LETs exposed ends and transcription initiation region. This modification combined with gel-based purification resulted in LETs expressing protein at equal activity as plasmids, reaffirming the results of previous work.
Conclusion
Use of LETs to express proteins at plasmid-levels without requiring the time-consuming steps of creating plasmid libraries (1/2 weeks) is a tremendous advantage for those who wish to screen hundreds of protein candidates while minimizing costs. Here we present some adjustments to conventional methods for of employing LETs in CFPS reactions that increase protein yield while maintaining their high-throughput advantages. I would like to thank Alan Harker’s generous sponsorship of the ORCA award that supported this project. I would like to offer a special thanks to Dr. Bradley Bundy and PhD student Matt Schinn whose mentoring was critical to this project’s success.