
Matt Phillips and Dr. William McCleary, Microbiology and Molecular Biology
I. Introduction
The ability to control gene expression in bacteria has been essential in solving problems in many fields, including medicine and environmental protection. Recent advances in genomic and metabolic modeling tools have led to the development of a new technique called promoter swapping which enables researchers to “swap” any native gene promoter with one that has been specifically engineered. Promoter swapping uses viral recombination proteins to swap DNA in the chromosome rather than in plasmids, allowing for constant and permanent expression levels. Our goal was to create eight template plasmids with promoters of increasing strengths to be used in promoter swapping. During the project, we made two plasmids with distinct promoters, verified their sequences, and successfully swapped one of them with the β-galactosidase promoter of E. coli. Thus, we created the first of eight template plasmids to be used in promoter swapping. Future researchers of this project will hopefully create the remaining seven plasmids by adjusting the sequence of the plasmid.
II. Methodology
A. Cloning of plasmid pKE2 with promoter insert to create template plasmid
First, we created a promoter sequence with all the necessary sequence motifs and three unique restriction enzyme sites. We ordered the sequence in four overlapping segments from Invitrogen. After the segments were annealed, we digested plasmid pKE2 with restriction enzymes, EcoRI and NdeI, to create a linear vector. This plasmid contained DNA regions necessary for cloning and swapping: the upstream and downstream homology regions to target the β-galactosidase promoter, the kanamycin-resistance (Kmr) cassette to select for the promoter, and the restriction enzyme sequences to facilitate cloning with the template plasmid. We also treated the vector with Antarctic Phosphatase to remove 5’ phosphates to increase efficiency of the cloning. Then, we ligated the insert with the digested vector to create the complete template plasmid.
B. Electroporation of the template plasmid into strain BW25141
We electroporated the template into E. Coli strain BW25141 and plated the culture onto Luria-Bertani (LB) broth and kanamycin plates to select for colonies with the template plasmid. We included a no- DNA control to check effectiveness of the cloning. The plates were grown for ~24 hours at 37°C, and several colonies were transferred to 5 mL of LB to grow overnight cultures. We isolated plasmids from each of the colonies with the Zymo Research Zyppy Plasmid Miniprep Kit. Next, we performed various restriction enzyme digests (i.e., Kpn1, BamHI, and EcoRV) with negative controls to test for the presence of the insert. Samples of each digest were run on 0.4% agarose gels, and two samples that displayed correct digestion were chosen for sequencing analysis and named pMP1-1 and pMP1-2. Sequencing results showed that pMP1-2 contained the exact sequence we created, while pMP1-1 had a single base pair mutation in the -35 region.
C. Electroporation of the plasmid PCR products into strain MG1655 + pKD46
With the sequences verified, we created PCR products of pMP1-1 and pMP1-2 to electroporate into a strain expressing the phage λ Red recombination proteins, MG1655 + pDK46. This was done using the New England Bio Labs Phusion Polymerase Kit. In order to increase DNA concentration, we performed duplicate PCRs and combined them before purification with the Qiagen PCR Purification Kit. We ran an agarose gel to confirm the PCR products were the right length and then determined the oligonucleotide concentration by Nano-drop. Next, we electroporated the PCR Activity units products into MG1655 + pKD46. Then, we plated 0.5 mL of the culture onto LB plates with kanamycin at 37°C. When there was no growth, we plated the remaining 0.5 mL after 24 hours of rest at room temperature. Colonies were obtained on both pMP1-1 and pMP1-2 samples.
D. Whole-colony PCR verification and Removal of the Kanamycin resistance gene
In order to confirm that our PCR products had been incorporated into the MG1655 genome, one colony from pMP1-1 and pMP1-2 was streaked for isolation on LB/Kan plates and grown at 42°C to remove pKD46 (a temperature-sensitive plasmid). Then, whole-colony PCR was completed with two primers, lacfor and k1, to generate a 700 base pair band and verify the presence of the DNA insert. Both pMP1-1 and pMP1-2 showed positive PCR tests, so overnight cultures with kanamycin in 5 mL LB were set up with a colony from each strain and used to make competent cells. We transformed each strain with pCP20, a temperature sensitive plasmid expressing the FLP recombinase system, to remove the Kmr cassette. We plated transformed cells onto LB plates with ampicillin and incubated them for ~24 hours at 30°C. Five colonies were selected from each strain, streaked for isolation on LB plates, and grown overnight at 42°C to remove pCP20. One colony from each plate was patched onto LB, LB/Kan, and LB/Amp plates and incubated overnight at 37°C.
E. β-galactosidase assays
One colony from each strain that simultaneously lost Kan and Amp resistance (i.e., removal of the antibiotic cassette and loss of pCP20) was used to grow overnight cultures for β-galactosidase assays. We grew six cultures of each strain as well as six overnight cultures of MG1655, the wild type. IPTG, a gratuitous inducer of the native β-galactosidase promoter, was added to half of the cultures before incubation. Cultures were assayed for β-galactosidase activity, which indirectly shows the activity of each promoter by measuring how much β-galactosidase was expressed.
III. Results
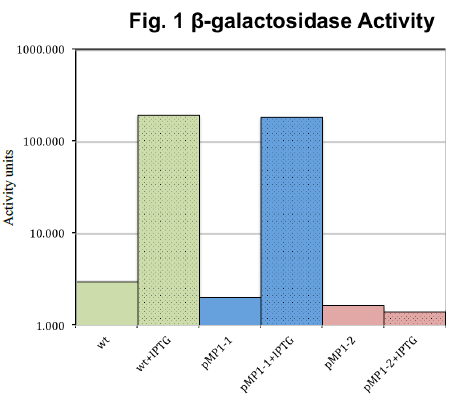
Our results are summarized in Fig. 1. Expression of β-galactosidase with the promoter from pMP1- 2 (red) was independent of IPTG, but expression for the pMP1-1 promoter (blue) was almost identical to wild type. Wild type (green) responded as expected, increasing β- galactosidase more than 60-fold with addition of IPTG.
IV. Discussion
First, the independence of pMP1-2 from IPTG is evidence that we successfully swapped the native β-galactosidase promoter with our synthetic promoter. This trend was confirmed in three consecutive β-galactosidase assays. However, the low expression level of pMP1-2 contradicts our hypothesis that a standard synthetic promoter would have high basal expression levels. Regarding pMP1-1, it behaved almost identically to the wild type with IPTG addition, which suggests that the native promoter was in fact not switched. It is unclear how mPM1-1 had a positive PCR test if the native promoter was not switched.
V. Conclusion
Though we were only able to make one of the eight proposed plasmids, our work paved the way for other researchers in Dr. McCleary’s lab to continue the project. We found it is difficult to predict promoter strength based only on the nucleotide sequence, so future projects will likely include mutagenesis of pMP1-2 to select for varying levels of expression. Depending on the outcome, this project could lead to a publication and a patent on the plasmids. Further, future experiments using our template plasmids could lead to important discoveries in bacterial physiology and biotechnology.