
David Broadbent, Dr. Joshua Andersen, Department of Chemistry and Biochemistry
The resistance of tumor cells to chemotherapy is a critical problem in the clinic and is a common cause of mortality in cancer. Emerging data suggests that resistance to chemotherapy is caused by a tumor-expressed protein called 14-3-3ζ, yet the mechanism to explain 14-3-3ζ-mediated chemoresistance is not completely understood (1). Rapid growing tumors often outgrow their blood supply resulting in regions of hypoxia (low glucose and oxygen). These cells must adapt or die and those that do adapt often become metastatic and chemoresistant. A recently proposed mechanism by which cancer cells are able to adapt to hypoxia is via autophagy, a process in which tumor cells recycle their own components for energy. My research with Dr. Andersen has uncovered a molecular pathway by which 14-3-3ζ promotes autophagy by driving the formation of lipid-enclosed vesicles called autophagosomes, the core cellular structures that recycle cellular contents for energy during periods of hypoxic stress. Specifically, we found that 14-3-3ζ accomplishes this by interacting with a protein called Autophagy related protein 9 (Atg9) which promotes autophagosome formation by delivering golgi-derived membrane to budding autophagosomes. Our data were recently published as the cover story in the December issue of Molecular and Cellular Biology.
Prior to starting this project, our laboratory knew very little about Atg9 and had limited experience in the field of autophagy. After making the initial discovery that 14-3-3ζ was linked to Atg9 and autophagy, it became apparent that we had to quickly bring ourselves up to speed in this relatively new field. Mechanisms of autophagy and autophagy assays are being pioneered by only a handful of prominent labs throughout the world, one of which is the laboratory of Dr. Sharon Tooze at the London Research Institute. Therefore, last Winter Dr. Andersen contacted Dr. Sharon Tooze to try to arrange an internship for me in her lab. She responded graciously and agreed to allow me to work in her lab with an autophagy research team over the summer to learn commonly used strategies and techniques in the field.
Working in her lab was one of my favorite educational experiences. I worked directly under PhD student Hannah Dooley, but was able to spend significant time being mentored by Dr. Tooze and the rest of her lab. I was very impressed by their expertise and how friendly they were. I performed many autophagy experiments with a focus on confocal microscopy. I was able to bring these techniques back to the Andersen lab and we now use them routinely in our 14-3-3ζ-Atg9 project.
Over the last year, we discovered that the interaction between 14-3-3ζ and Atg9 is dependent on Atg9 phosphorylation at Serine 761. One can think of this phosphorylation as an on-switch for Atg9 activity. In order to study this phosphorylation in detail, we generated a phosphorylation-resistant RFP-tagged mutant of Atg9 that we refer to as S761A. Importantly, this S761A mutant Atg9 is unable to interact with 14-3-3ζ. One way to assess Atg9 activity is by monitoring its trafficking between golgi and autophagosomes. Using the confocal techniques I learned in the Tooze lab, I have monitored the interaction of Atg9 with a protein called DFCP1, which is a marker of early autophagosomal precursors called omegasomes. Previous reseach has shown that cells depleted of Atg9 have reduced DFCP1 puncta and I hypothesized that 14-3-3ζ promotes Atg9 trafficking to DFCP1-positive omegasomes (2). My preliminary data below support this hypothesis. In addition, I found that cells expressing the S761A mutant Atg9 shower lower overall numbers of DFCP1 puncta, indicative of defective autophagy.
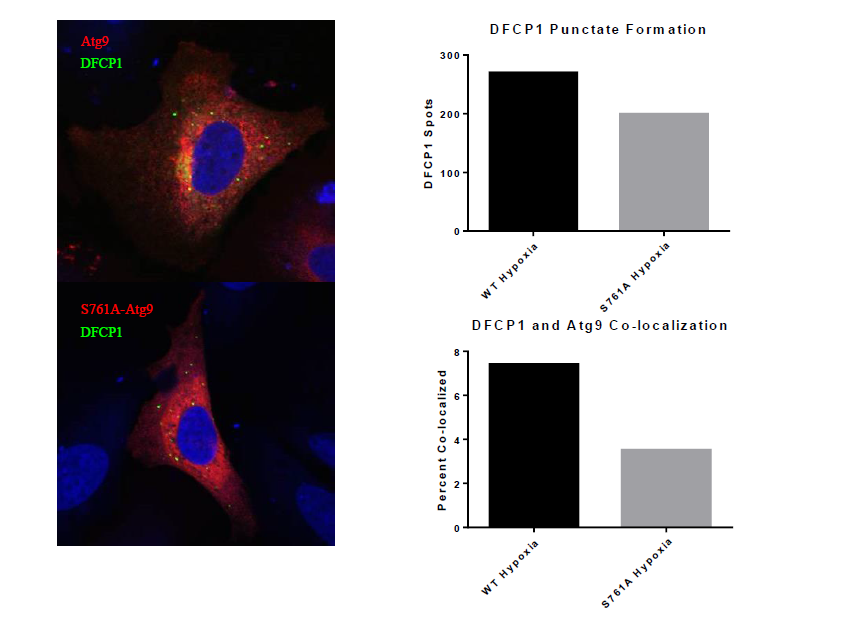
While these results are still preliminary, they suggest that in the absence of S761 phosphorylation and 14-3-3z binding, Atg9 is defective in localizing to the DFCP1-containing autophagosomal precursors. This is a major contribution to our understanding of autophagy regulation in cancer cells. In the big picture, our data suggest that by interfering with Atg9 phosphorylation at S761 we may be able to sensitize tumors to hypoxia and other stresses, such as chemotherapy, and ultimately enhance the eradication of tumor cells from the body.
1. Neal, C. L., and Yao J., et al. (2009) 14-3-3ζ Overexpression Defines High Risk for Breast Cancer Recurrence and Promotes Cancer Cell Survival. Cancer Res. 69(8):3425- 32
2. Orsi, A., Razi, M., Dooley, H. C., Robinson, D., Weston, A. E., Collinson, L. M., & Tooze, S. A. (2012). Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy.Molecular Biology of the Cell, 23(10), 1860–1873. doi:10.1091/mbc.E11-09-0746