
Jeffery Nielson and Dr. Alonzo Cook, Department of Chemical Engineering
Introduction
Several kinds of organ failure are among the leading causes of death in the United States. Organ donations cannot meet the demand for organ transplants due to the sensitive time period in which viable organs can be harvested and utilized. Those patients who actually do receive organ transplants (roughly 25% of those on the transplant waitlist) are required to take immunosuppressant drugs for the rest of their lives, in order to prevent their bodies from rejecting the transplanted organ. Not only is this a costly and lengthy process of waiting for the right donor, but it also results in a dangerous situation for the patients because their immune systems are weakened for the remainder of their lives. One alternative approach to meeting the demand for organ transplants is to grow organs in a lab, using the patient’s own cells on an organ scaffold. Some have used synthetic scaffolding for the patient’s cells to grow on. Another approach, however, is to decellularize organs that come from organ donors who have been deceased too long for their organs to be viable for direct transplant. Using a combination of surfactants and other solutions on these currently non-transplantable organs, all cells and any immunogenic cellular material can be removed from the organs, leaving an inert collagen scaffolding known as the Extracellular matrix (ECM). The ECM retains the growth factors and proteins necessary for cell growth and cell differentiation. However, many surfactants used in organ decellularization can denature proteins and therefore damage the integrity of the ECM as a scaffold for recellularization. This damage has resulted in failure for most attempts of in vitro whole organ growth and for all attempts to grow solid organs. The issue facing most researchers in the field of in vitro organ growth is figuring out how they can quantify the integrity of their decellularized ECMs, in order to assess the decellularization methods that they use. Fluorescence-based cell quantification is a possible method for ECM integrity quantification.
Methodology
The linear relationship of cell concentration to fluorescence was assessed using trypsin-detached mouse MS-1 (microvascular endothelial) cells at different concentrations. The different concentrations of cells were prepared by growing MS-1 in a T-75 flask in DMEM with 10% FBS and 1% Pen-Strep. The MS-1 cells were detached by trypsin and quantified by a Moxi-automated cell counter. The fluorescence of each concentration prepared in a 96-well plate was measured by using a fluorometer. The resulting correlation was analyzed. Two different decellularization methods were assessed, the first being the most common method of decellularization: continuous antegrade internal perfusion of 0.5% sodium dodecyl sulfate (SDS) solution. The other method of decellularization is known as tonic cycles. Tonic cycles consist of alternating antegrade internal perfusion between a series of 3 different solutions: distilled water, 0.5M Saline, and 0.5% SDS solution. The resulting decellularized ECMs were sterilized by incubation in a solution of 1000 mg/mL streptomycin, 1000 U/mL penicillin and 25 μg/mL
amphotericin B in 70% ethanol at room temperature for 2 h with proper agitation. The biocompatibility of the decellularized ECM was confirmed by cell adhesion. The kidneys were dissected and a 7 mm diameter circular section was taken from the cortex. Then, the samples were soaked twice in PBS and incubated in cell growth medium containing DMEM with 10% FBS and 1% Pen-Strep at 37°C and 5% CO2 in 24-well plates. The growth medium was replaced after 2 days and trypsin-detached mouse MS-1 cells (1×105) were added to each well. The growth medium was changed every 2 days and after 3 days of cell culture, tissue samples were taken out of the wells, fixed in 4% paraformaldehyde for 24 h and prepared for sectioning and histology. The survival of the cells that came in contact with the kidney tissue samples were assessed using a Biotium® Viability/Cytotoxicity Assay, which was used after the samples were taken out of the culture well plates. Viable cells were marked with fluorescence (see results in Figure 2). The fluorescence of the cells was measured using a FLoid® Cell Imaging Station.
Results
The correlation of measured fluorescence-to-cell concentration only had an R squared value of 0.75. The result of recellularization attempts are listed below in Figure 1. The results of cell viability tests are listed in Figure 2.
Discussion
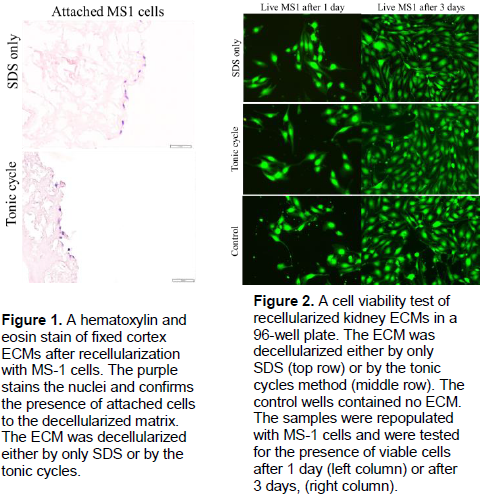
The poor R2 value in the correlation between cell concentration and fluorescence is indicative of a non-linear relationship between cells and the amount of fluorescence. Potential causes of this poor correlation include cells varying in size and the amount of GFP produced in each cell not being a fixed constant. This nullifies any quantitative measurements of cells made by fluorescence alone. The fluorescence can be used to confirm the presence of cells, however, other methods had to be used to confirm cell viability. Both decellularization methods produced non-cytotoxic scaffolds that can support recellularization and cell proliferation. However, the scaffolds only allowed for cells to attach on the outer surface and in the vasculature.
Conclusion
Fluorescence-based cell adhesion testing does not allow for any insight into the integrity of the ECM, because there is not a linear correlation between fluorescence and cell quantity. Other methods helped confirm the viability of these scaffolds in supporting recellularization, but did not illuminate the differences in ECM preservation between different methods. Alternative methods for ECM integrity testing should be the subject of future research.