
Mitchell Gleed and Professor David D. Busath, M.D., Physiology and Developmental Biology
INTRODUCTION
Influenza is responsible for millions of hospitalizations and deaths every year. Treating the influenza virus has become increasingly difficult since mutant viruses have rendered previously viable drugs ineffective. New drug-screening procedures are now necessary in order to speed up the process of viable drug discovery. Molecular simulations have proven valuable for studying drug effectiveness in wild-type flu viruses, but little has been done on prevailing influenza mutants. Influenza mutants often feature a change in the M2 proton channel, a critical component in the viral replication and infection cycle. To counter this, drugs must be discovered that can block the influenza M2 channel of mutant viruses. The purpose of this research report is to discuss the methods and results of using molecular dynamics simulations to predict drug efficacy against the M2 proton channel of the Influenza A virus.
METHODOLOGY
Using an NMR model of wild-type M2, binding affinity (which governs drug entry to the binding site) of adamantane compounds for the M2 drug-binding site and water (representing extra-viral fluid) was be assayed in CHARMM. By comparing binding affinity between these locations, drug efficacy can be measured and predicted. To achieve this end, we developed a method which employs TSM perturbation within CHARMM, a molecular dynamics software package. TSM perturbation allows for Gibb’s free energy (ΔG) to be calculated by comparing different drugs in the same environment, rather than comparing the one drug in different environments. Free-energy perturbation allows for background free-energy variance such as that produced by water molecules to be greatly reduced and be prevented from interfering with data of interest. Knowing the Gibbs free energy of each drug in each environment enables us to evaluate the viability of various drugs in the M2 channel. We began by creating 20 drugs in ChemBio3D and prepared them for use in simulations using swissparam.ch. Once the drugs were created, we downloaded a solid-state NMR structure of wild-type M2 from the Protein Databank (PDBID 2KQT), prepared it for use in CHARMM, placed it in a lipid bilayer, hydrated the channel, and then placed the drugs into the channel. We then heated up the systems to 310 degrees Kelvin and allowed the systems to equilibrate for up to 10.5 nanoseconds of simulation time. Light constraints were applied to the M2 channel in order to keep it near the average configuration of all of the NMR models given from pdb.org.
RESULTS
After the simulations completed, we analyzed the TSM perturbation output files and gathered ΔG’s of each of the drugs. Figure 1a shows the ΔG of each drug as they were mutated piece-wise via the TSM method at various degrees (lambda) in the M2 binding site.
We also gathered ΔG’s of the drugs as they were mutated with TSM at varying lambdas. We subtracted these ΔG’s from the ΔG’s observed in the M2 proton channel to get ΔΔG’s, which we could then use to compare to experimental results. This comparison is illustrated in Figure 1b, which plots experimental ΔΔG’s against TSM ΔΔG’s.
DISCUSSION
We are pleased to see consistency with ΔG’s between both the M2 channel simulations and the simulations with drugs in only water. These results show that some drugs tend to have higher binding affinity while others had lower binding affinity for the M2 proton channel than Amantadine, a potent drug that blocks wild-type influenza.
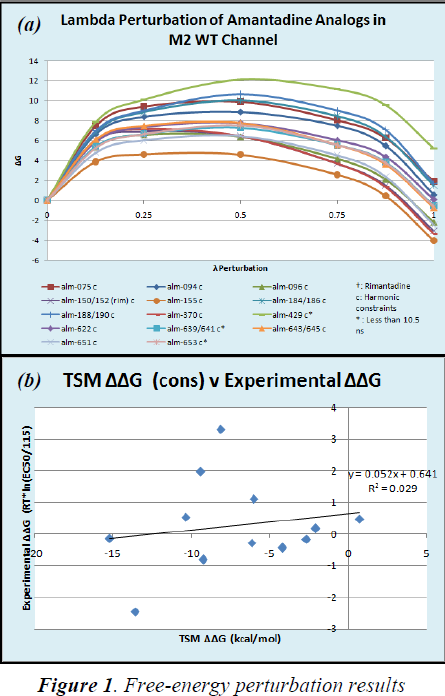
Our ΔΔG’s as determined by the TSM method did not correlate well with experimental results as shown in Figure 2. The low R2 value of 0.029 and the low slope value of 0.052 both indicate a very poor correlation between data.
CONCLUSIONS
Due to the poor correlation we observed between our TSM perturbation data and experimental data, we concluded that we should not go on to apply this method to M2-channel mutants without first revising our procedures and performing simulations again. We are, however, encouraged by the consistent results we received for measuring ΔG’s, which illustrates to us that the TSM technique is self-consistent, and that we merely need to make adjustments to our procedure and methods to get more reliable results.
In future experiments we plan to use a newer version of CHARMM, including a newer force field with newer, more-reliable parameters for atomic interactions. We also plan to repeat each simulation with different initial velocities to give average ΔG’s with standard errors, to ensure that we are getting reliable data from TSM. Furthermore, we will increase our lambda schedule from three to seven values of lambda in order to provide better approximations of ΔG near the endpoints. With these adjustments and more, we are confident that we will be able to apply free-energy perturbation methods to reflect drug efficacy measured by in-vitro assays.